
 | ||
Nucleation is the first step in the formation of either a new thermodynamic phase or a new structure via self-assembly or self-organisation. Nucleation is typically defined to be the process that determines how long we have to wait before the new phase or self-organised structure appears. Classical nucleation theory (CNT) is the most common theoretical model used to understand why nucleation may take hours or years, or in effect never happen.
Contents
Outline of classical nucleation theory
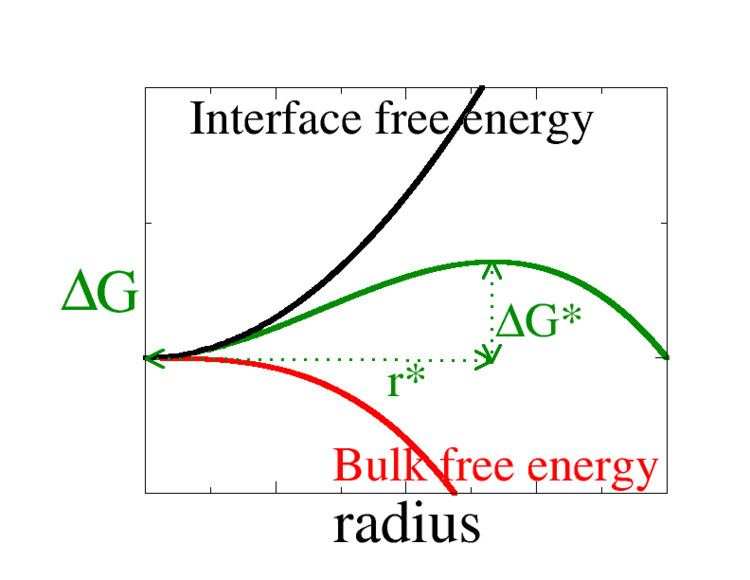
This is the standard simple theory for nucleation of a new thermodynamic phase, such as a liquid or a crystal. It should be borne in mind that it is approximate. The basic CNT nucleation of a new phase provides an approximate but physically reasonable prediction for the rate at which nuclei of a new phase form, via nucleation on a set of identical nucleation sites. This rate, R is the number of, for example, water droplets nucleating in a uniform volume of air supersaturated with water vapour, per unit time. So if a 100 droplets nucleate in a volume of 0.1m3 in 1s, then the rate R=1000/s. The description here follows modern standard CNT. The prediction for the rate R is
where
This expression for the rate can be thought of as a product of two factors: The first,
The second factor in the expression for the rate is the dynamic part,
To see how this works in practice we can look at an example. Sanz and coworkers have used computer simulation to estimate all the quantities in the above equation, for the nucleation of ice in liquid water. They did this for a simple but approximate model of water called TIP5P/2005. At a supercooling of 19.5 °C, i.e., 19.5 °C below the freezing point of water in their model, they estimate a free energy barrier to nucleation of ice of
This is a rate of homogeneous nucleation estimated for a model of water, not real water—in experiments we cannot grow nuclei of water and so cannot directly determine the values of the barrier ΔG*, or the dynamic parameters such as j, for real water. However, it may be that indeed the homogeneous nucleation of ice at temperatures near -20 °C and above is extremely slow and so that whenever we see water freezing temperatures of -20 °C and above this is due to heterogeneous nucleation, i.e., the ice nucleates in contact with a surface.
Homogeneous nucleation
Homogeneous nucleation is much rarer than heterogeneous nucleation. However, homogeneous nucleation is simpler and easier to understand than heterogeneous nucleation, so the easiest way to understand heterogeneous nucleation is to start with homogeneous nucleation. So we will outline the CNT calculation for the homogeneous nucleation barrier
To understand if nucleation is fast or slow,
The first term is the volume term, and as we are assuming that the nucleus is spherical, this is the volume of a sphere of radius
For small
Addition of new molecules to nuclei larger than this critical radius decreases the free energy, so these nuclei are more probable. The rate at which nucleation occurs is then limited by, i.e., determined by the probability, of forming the critical nucleus. This is just the exponential of minus the free energy of the critical nucleus
This is the free energy barrier needed in the CNT expression for
Heterogeneous nucleation
Heterogeneous nucleation, nucleation with the nucleus at a surface, is much more common than homogeneous nucleation. Heterogeneous nucleation is typically much faster than homogeneous nucleation because the nucleation barrier ΔG* is much lower at a surface. This is because the nucleation barrier comes from the positive term in the free energy ΔG, which is the surface term. For homogeneous nucleation the nucleus is approximated by a sphere and so has a free energy equal to the surface area of a sphere, 4πr2, times the surface tension σ. However, as we can see in the schematic of macroscopic droplets to the right, droplets on surfaces are not complete spheres and so the area of the interface between the droplet and the surrounding fluid is less than
In the schematic to the right the contact angle between the droplet surface and the surface decreases from left to right (A to C), and we see that the surface area of the droplet decreases as the contact angle decreases. This geometrical effect reduces the barrier and hence results in faster nucleation on surfaces with smaller contact angles. Also, if instead of the surface being flat it curves towards fluid, then this also reduces the interfacial area and so the nucleation barrier. There are expressions for this reduction for simple surface geometries. In practice, this means we expect nucleation to be fastest on pits or cracks in surfaces made of material such that the nucleus forms a small contact angle on its surface.
Comparison between CNT and computer simulation
The classical nucleation theory makes a number of assumptions, for example it treats a microscopic nucleus as if it is a macroscopic droplet with a well defined surface whose free energy is estimated using an equilibrium property: the interfacial tension σ. For a nucleus that may be only of order ten molecules across it is not always clear that we can treat something so small as a volume plus a surface. Also nucleation is an inherently out of thermodynamic equilibrium phenomenon so it is not always obvious that its rate can estimated using equilibrium properties.
However, modern computers are powerful enough to calculate essentially exact nucleation rates, but only for simple models. These have been compared with the classical theory, for example for the case of nucleation of the crystal phase in the model of hard spheres. This is a model of perfectly hard spheres in thermal motion, and is a simple model of some colloids. For the crystallization of hard spheres the classical theory is a very reasonable approximate theory. So for the simple models we can study CNT works quite well, but we do not know if it works equally well for say complex molecules crystallising out of solution.