
 | ||
Carbohydrate synthesis is a sub-field of organic chemistry concerned specifically with the generation of natural and unnatural carbohydrate structures. This can include the synthesis of monosaccharide residues or structures containing more than one monosaccharide, known as oligosaccharides.
Contents
Background
Generally speaking, carbohydrates can be classified into two groups, simple sugars and complex carbohydrates. Simple sugars, also called monosaccharides, are carbohydrates which can not be converted into smaller sugars by hydrolysis. When two or more monosaccharide units are connected to one another via a glycoside linkage, complex carbohydrates are formed. Complex carbohydrates, according to the different number of monosaccharide units, can be classed into three groups, disaccharides, oligosaccharides, and polysaccharides. A disaccharide is formed from two monosaccharides. Oligosaccharides can be formed by a small number of monosaccharides linked together. Higher oligosaccharides are called polysaccharides. It is now well known that glycoconjugates play an indispensable role in many biological processes. These biological processes in which carbohydrates are involved are typically associated not to monosaccharides, but to oligosaccharides structures of glycoconjugates. Therefore, the oligosaccharide synthesis becomes more and more important in studying the biological activities.
Oligosaccharide synthesis
Oligosaccharides have diverse structures. The number of monosaccharides, ring size, the different anomeric stereochemistry, and the existence of the branched-chain sugars all contribute to the amazing complexity of the oligosaccharide structures. The essence of the reducing oligosaccharide synthesis is connecting the anomeric hydroxyl of the glycosyl donors to the alcoholic hydroxyl groups of the glycosyl acceptors. Protection of the hydroxyl groups of the acceptor with the target alcoholic hydroxyl group unprotected can assure the regiochemical control. Additionally, factors such as the different protecting groups, the solvent, and the glycosylation methods can influence the anomeric configurations. This concept is illustrated by an oligosaccharide synthesis in Scheme 1. Oligosaccharide synthesis normally consists of four parts: preparation of the glycosyl donors, preparation of the glycosyl acceptors with a single unprotected hydroxyl group, the coupling of them, and the deprotection process.
Building blocks
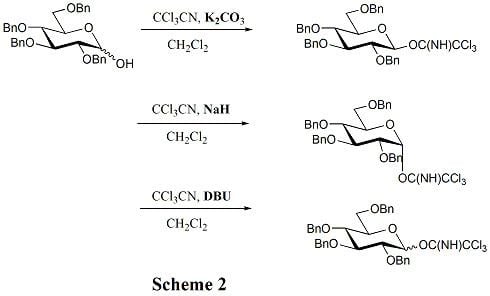
Common donors in oligosaccharide synthesis are glycosyl halides, glycosyl acetates, thioglycosides, trichloroacetimidates, pentenyl glycosides, and glycals. Of all these donors, glycosyl halides are classic donors, which played a historical role in the development of glycosylation reactions. Thioglycoside and trichloroacetimidate donors are used more than others in contemporary glycosylation methods. When it comes to the trichloroacetimidate method, one of the advantages is that there is no need to introduce heavy metal reagents in the activation process. Moreover, using different bases can selectively lead to different anomeric configurations. (Scheme 2) As to the thioglycosides, the greatest strength is that they can offer a temporary protection to the anomeric centre because they can survive after most of the activation processes. Additionally, a variety of activation methods can be employed, such as NIS/ AgOTf, NIS/ TfOH, IDCP (Iodine dicollidine perchlorate), iodine, and Ph2SO/ Tf2O. Furthermore, in the preparation of 1, 2-trans glycosidic linkage, using thioglycosides and imidates can promote the rearrangement of the orthoester byproducts, since the reaction mixtures are acidic enough.
Stereoselectivity
The structures of acceptors play a critical role in the rate and stereoselectivity of glycosylations. Generally, the unprotected hydroxyl groups are less reactive when they are between bulky protecting groups. That is the reason why the hydroxyl group at OH-4 in pyranosides is unreactive. Hyperconjugation is involved when OH-4 is anti-periplanar to the ring oxygen, which can also reduce its reactivity. (Scheme 3) Furthermore, acyl protecting groups can reduce the reactivity of the acceptors compared with alkyl protecting groups because of their electron-withdrawing ability. Hydroxyl group at OH-4 of N-acetylglucosamine derivatives is particularly unreactive.
The glycosidic bond is formed from a glycosyl donor and a glycosyl acceptor. There are four types of glycosidic linkages: 1, 2-trans-α, 1, 2-trans-beta, 1, 2-cis-α, and 1, 2-cis-beta linkages. 1, 2-trans glycosidic linkages can be easily achieved by using 2-O-acylated glycosyl donors (neighboring group participation). To prevent the accumulation of the orthoester intermediates, the glycosylation condition should be slightly acidic.
Difficult linkages
It is somewhat more difficult to prepare 1, 2-cis-α glycosidic linkages stereoselectively. Typically, when non-participating groups on O-2 position, 1, 2-cis-α linkage can be achieved either by using the historically important halide ion methods, or by using 2-O-alkylated glycosyl donors, commonly thioglycosides or trichloroacetimidates, in nonpolar solvents.
In the early 1990s, it was still the case that the beta mannoside linkage was too challenging to be attempted by amateurs. However, the method introduced by Crich (Scheme 4), with 4,6-benzylidene protection a prerequisite and anomeric alpha triflate a key intermediate leaves this problem essentially solved. The concurrently developed but rather more protracted intramolecular aglycon delivery (IAD) approach is a little used but nevertheless stereospecific alternative.