
Specialty endocrinology ICD-9-CM 330.0 DiseasesDB 29780 | ICD-10 E75.2 OMIM 271900 MedlinePlus 001586 | |
 | ||
Canavan disease, also called Canavan–van Bogaert–Bertrand disease, is an autosomal recessive degenerative disorder that causes progressive damage to nerve cells in the brain, and is one of the most common degenerative cerebral diseases of infancy. It is caused by a deficiency of the enzyme aminoacylase 2, and is one of a group of genetic diseases referred to as leukodystrophies. It is characterized by degeneration of myelin in the phospholipid layer insulating the axon of a neuron and is associated with a gene located on human chromosome 17.
Contents
Symptoms
Symptoms of Canavan disease, which appear in early infancy usually between the first three to six months of age. Canavan disease then progresses rapidly from that stage, the following may include intellectual disability, loss of previously acquired motor skills, feeding difficulties, abnormal muscle tone (i.e., floppiness or stiffness), poor head control, and megalocephaly (abnormally enlarged head). Paralysis, blindness, or seizures may also occur.
Pathophysiology
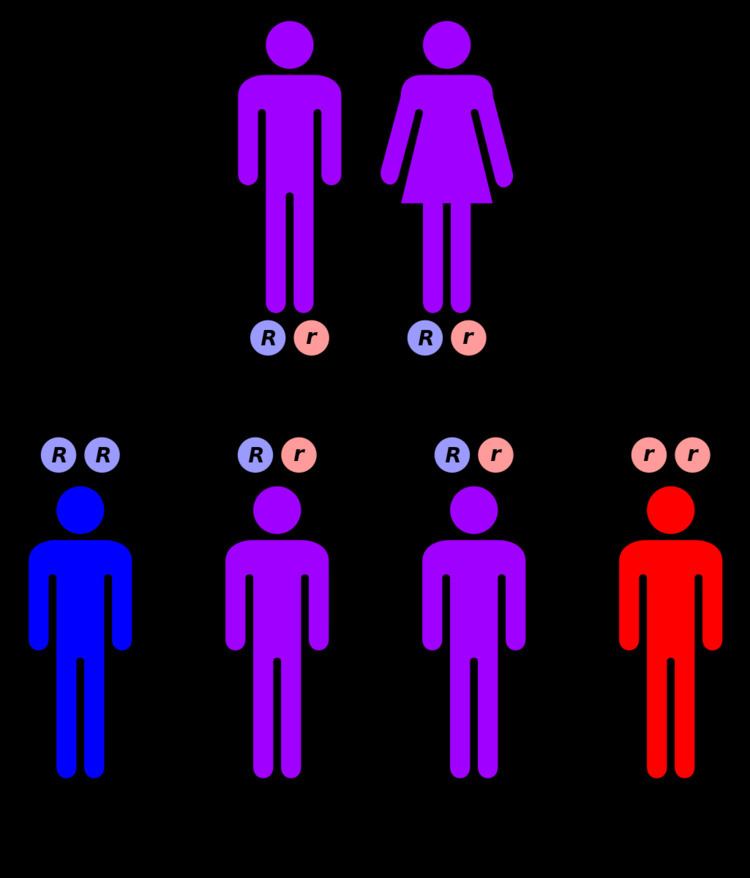
Canavan disease is inherited in an autosomal recessive fashion. When both parents are carriers, there is a 25% chance of having an affected child. Genetic counseling and genetic testing is recommended for families with two parental carriers.
Canavan disease is caused by a defective ASPA gene which is responsible for the production of the enzyme aspartoacylase. Decreased aspartoacylase activity prevents the normal breakdown of N-acetyl aspartate, wherein the accumulation of N-acetylaspartate, or lack of its further metabolism interferes with growth of the myelin sheath of the nerve fibers of the brain. The myelin sheath is the fatty covering that surrounds nerve cells and acts as an insulator, allowing for efficient transmission of nerve impulses.
Treatment
There is no cure for Canavan disease, nor is there a standard course of treatment. Treatment is symptomatic and supportive. There is also an experimental treatment using lithium citrate. When a person has Canavan disease, his or her levels of N-acetyl aspartate are chronically elevated. The lithium citrate has proven in a rat genetic model of Canavan disease to be able to significantly decrease levels of N-acetyl aspartate. When tested on a human, the subject's condition reversed during a two-week wash-out period after withdrawal of lithium.
The investigation revealed both decreased N-acetyl aspartate levels in regions of the brain tested and magnetic resonance spectroscopic values that are more characteristic of normal development and myelination. This evidence suggests that a larger controlled trial of lithium may be warranted as supportive therapy for children with Canavan disease.
Experimental gene therapy trial results, published in 2002, used a healthy gene to take over for the defective one that causes Canavan disease. In human trials, the results of which were published in 2012, this method appeared to improve the life of the patient without long-term adverse effects during a 5-year follow-up.
Prognosis
Death usually occurs before age ten, but some children with milder forms of the disease survive into their teens and twenties.
Prevalence
Although Canavan disease may occur in any ethnic group, it affects people of Eastern European Jewish ancestry more frequently. About 1 in 40 (2.5%) individuals of Eastern European (Ashkenazi) Jewish ancestry are carriers.
Research
Research involving triacetin supplementation has shown promise in a rat model. Triacetin, which can be enzymatically cleaved to form acetate, enters the brain more readily than the negatively charged acetate. The defective enzyme in Canavan disease, aspartoacylase, converts N-acetylaspartate into aspartate and acetate. Mutations in the gene for aspartoacylase prevent the breakdown of N-acetylaspartate, and reduce brain acetate availability during brain development. Acetate supplementation using Triacetin is meant to provide the missing acetate so that brain development can continue normally.
A team of researchers headed by Paola Leone at the University of Medicine and Dentistry of New Jersey, has trialed a procedure involving the insertion of six (6) catheters into the brain that deliver a solution containing 600 billion to 900 billion engineered virus particles. The virus, a modified version of AAV, is designed to replace the aspartoacylase enzyme. Children treated with this procedure to date have shown marked improvements, including the growth of myelin, with decreased levels of the n-acetyl-aspartate toxin.
History
Canavan disease was first described in 1931 by Myrtelle Canavan. In 1931, she co-wrote a paper discussing the case of a child who had died at sixteen months and whose brain had a spongy white section. Canavan was the first to identify this degenerative disorder of the central nervous system, which was later named "Canavan Disease."
Greenberg v. Miami Children's Hospital Research Institute
The discovery of the gene for Canavan disease, and subsequent events, generated considerable controversy. In 1987 the Greenbergs, a family with two children affected by Canavan disease, donated tissue samples to Reuben Matalon, a researcher at the University of Chicago who was looking for the Canavan gene. He successfully identified the gene in 1993 and developed a test for it that would enable antenatal (before birth) counseling of couples at risk of having a child with the disease. For a while, the Canavan Foundation offered free genetic testing using Matalon's test.
However, in 1997, after he relocated to Florida, Matalon's new employer, Miami Children's Hospital, patented the gene and started claiming royalties on the genetic test, forcing the Canavan Foundation to withdraw their free testing. A subsequent lawsuit brought by the Canavan Foundation against Miami Children's Hospital, and was resolved with a sealed out-of-court settlement. The case is sometimes cited in arguments about the appropriateness of patenting genes.