
Acronym CAPP-Seq Related items | Inventor Aaron M Newman et al. | |
 | ||
Notable experiments CAPP-Seq was applied on non–small-cell lung cancer (NSCLC) to identify recurrent somatic alterations from ctDNA. | ||
CAncer Personalized Profiling by deep Sequencing (CAPP-Seq) is a sensitive method used to quantify DNA in cancer. It measures Cell-free tumor DNA which is released from dead tumor cells into the blood. This method can be generalized for any cancer type that is known to have recurrent mutations. CAPP-Seq can detect one molecule of mutant DNA in 10,000 molecules of healthy DNA.
Contents
- Methods
- ctDNA Extraction and Library Preparation
- Selector design
- Hybridization Capture and Sequencing
- Computational pipeline for CAPP seq
- Sensitivity
- Limitations
- Advantages
- Monitoring tumor burden
- Prognostic indicator
- Biopsy free tumor genotyping
- References
CAPP-Seq was designed to lower sequencing costs by only targeting specific areas of the genome that are recurrently mutated for a given cancer. This allows for sequencing costs between $200-$300 USD. It can also target multiple areas of the genome at once and a variety of different types of mutations, allowing for a lower amount of input DNA compared to other methods.
Methods
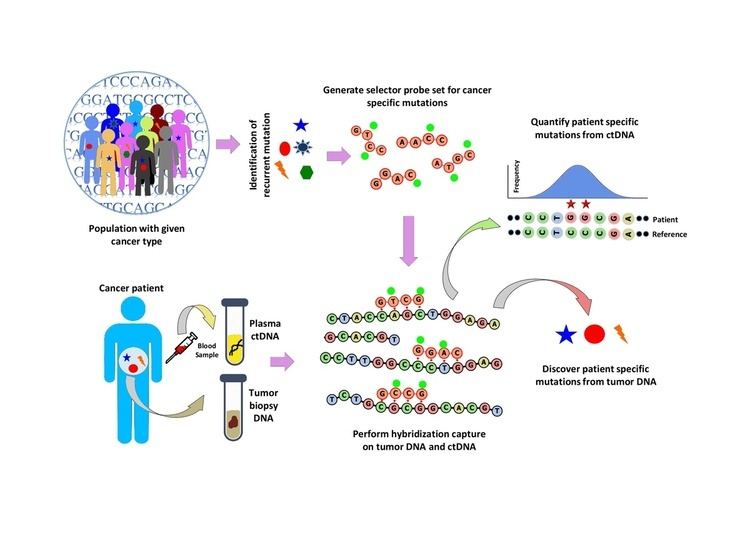
Population analysis is performed to identify recurrent mutations in a given cancer type. This is done by analyzing public data sets such as the COSMIC cancer database and TCGA. A ‘selector’ is designed which consists of biotinylated DNA oligonucleotide probes targeting the recurrently mutated regions chosen for the specific cancer type. The selector is chosen using a multiphase bioinformatics approach. Using the selector, a probe-based hybridization capture is performed on tumor and normal DNA to discover mutations specific to the patient. The hybridization capture is then also applied to ctDNA to quantify the mutations that were previously discovered.
ctDNA Extraction and Library Preparation
Peripheral blood is collected from patients and ctDNA is isolated from ≥1 mL of plasma. DNA libraries are made using the KAPA Library kit with some modifications in order to optimize protocols for use with ctDNA. The KAPA HiFi polymerase is used here because it is the most effective and produces the least amount of errors. Input DNA can be as low as 4 ng.
There were four main goals in adapting this protocol for ctDNA work:
These were achieved by allowing adaptor ligation to be carried out at 16℃ for 16 hours to increase adaptor ligation efficiency and recovery. The most important adaptation is during enzymatic and clean-up steps; they are performed with-bead, in order to minimize tube transfer steps which increases recovery.
Selector design
In CAPP-Seq, design of selector is a crucial step that identifies recurrent mutations in a particular cancer type using publicly available next generation sequencing data. For inclusion in CAPP-seq selector, the recurrent mutations that are enriched in a population is described by an index- Recurrence Index (RI). RI is the number of mutations per kilobase of a given genomic locus of a patient carrying particular mutations. RI represents a patient level recurrence frequency estimated for somatic mutations and all mutations. Known and driver recurrent mutations in a population can be ranked based on the RI and therefore RI is used to design a selector. A six phase design strategy is employed to design selector.
Human cancer is heterogeneous and recurrent cancer mutations are present only a minority of patient. Therefore, a careful and non-redundant design of selector is the vital part in CAPP-Seq and also the size of the selector is related to its downstream costs.
Hybridization Capture and Sequencing
Hybridization capture with the selector probe set is performed on tumor DNA from a biopsy and sequenced to a depth of ~10,000× coverage. The biotinylated selector probes bind selectively to the regions of the DNA library that were chosen to be where the recurrently mutations occur in the given cancer type. In this way you are left with a smaller library that is enriched for only the regions you want, which can then be sequenced. This allows the determination of patient specific mutations. Hybridization capture with the same selector is then performed on ctDNA from the blood to quantify the previously identified mutations in the patient. CAPP-Seq can be applied to ctDNA from multiple blood samples at different time points in order to follow tumor evolution.
Computational pipeline for CAPP-seq
A series of steps are involved in analysis of CAPP-Seq data from mutation detection to validation and open source software can do most of the analysis. After the first step of variant calling, germline and loss of heterozygosity (LOH) mutations are removed in CAPP-seq to reduce the background biases. Several statistical significance tests can be performed against background to all type of variant calling. For example, statistical significance of tumor-derived SNVs can be estimated by random sampling of background alleles using Monte Carlo method. For the indel calls, statistical significance is calculated applying a separate method that used a strand specific analysis by Z-test shown in previous work. Finally, a computational validation steps reduces the false positive calls. However, a robust computational framework specific for CAPP-seq data analysis is a high demand in this field.
Sensitivity
Sensitivity of this technology depends on the effective design of selector and highly biased with the size of the cohort and type of cancer under study. The lack of background to find the statistically significant recurrent variants has limited its performance due to stochastic noise and biological variability. Receiver Operating Characteristic (ROC) analysis on several cancer patient and cancer cured patient (sample collected at different tumor stages, circulating DNA time point, treatment, etc.) showed that CAPP-seq has higher sensitivity and specificity compared to previous methods in non–small-cell lung cancer.
Limitations
The detection limit of CAPP-Seq is affected by three main areas: the input amount of ctDNA molecules, sample cross-contamination, potential allelic bias in the capture reagent, and PCR or sequencing errors. CtDNA is able to be detected at a lower limit of 0.025% fractional abundance in the blood. Sample cross-contamination was found to be a very small contribution and reports have shown minimal allelic bias towards capture of reference alleles in PBLs (peripheral blood lymphocytes). PCR and sequencing errors are also minimal.
It is important to note that when using CAPP-Seq on ctDNA it still currently not known whether ctDNA is released at equal rates from primary tumors and metastatic disease, so this should be taken into consideration. This could cause problems with determining tumor burden and clonal evolution if different tumors or clones are dying off and releasing their DNA at different rates. It is also unknown how tumor histology affects ctDNA release.
Another major limitation with only using ctDNA levels to detect tumor burden is that ctDNA can only predict residual tumor, it cannot tell you where the tumor or tumors are located within the body. This means that ctDNA will be best used complementary to imaging for disease burden at this time.
Advantages
CAPP-Seq has many advantages over other methods such as digital polymerase chain reaction (dPCR) and amplicon sequencing. CAPP-Seq can survey many loci in the same experiment compared to dPCR and amplicon sequencing which use multiple different experiments and therefore use up much more sample. Another advantage is that CAPP-Seq can not only detect point mutations but it can also detect indels, structural variations, and copy number variations.
Another advantage of CAPP-Seq is that because it only targets specific areas of interest in the genome it is more cost effective than whole exome sequencing and whole genome sequencing which are 44X and 171X more expensive respectively.
Using circulating tumor DNA as opposed to solid tumor biopsies allows analysis of the full repertoire of tumor cells dispersed throughout the tumor and distant metastasis. Therefore, there is a better chance of finding all mutations associated with this cancer. Having a full overview of the cancer and what is driving it will allow for better treatment plans and management of disease.
Monitoring tumor burden
When treating cancer it is useful to have precise measurements of the total body disease burden. It helps with determining prognostic significance and treatment response. This is normally done using computed tomography (CT scans), positron emission tomography (PET scans) or magnetic resonance imaging (MRI). These medical imaging procedures are expensive and are not without their own problems. These imaging techniques are not able to accurately resolve small tumors (≤1 cm in diameter). Imaging can also be affected by radiation-induced inflammation and fibrotic changes, making is hard to determine if there is residual tumor or just effects of treatment.
It has been found that levels of ctDNA in plasma significantly correlate with tumor volume as compare with medical imaging (CT, PET and MRI)., Detection of ctDNA can predict residual tumor or imminent relapse, in some cases even better than medical imaging and current methods.
Prognostic indicator
Detection of ctDNA has been found to be a predictor of relapse in multiple studies thus far. In a study in late stage NSCLC (non-small cell lung cancer) they found two cases where ctDNA correctly determined the outcome of a patient when medical imaging was wrong. In one cause, the imaging predicted relapse based on a suspected residual tumor which turned out to only be radiation induced inflammation, but ctDNA was not detected and the patient did not relapse. In another case, the imaging showed no tumor but ctDNA was detected and the patient relapsed shortly afterwards. In another study on DLBCL (diffuse large B cell lymphoma), ctDNA was also found to be predictive of relapse.
Biopsy-free tumor genotyping
Biopsies are invasive and associated with risks to the patient. Therefore, multiple biopsies to monitor disease progression are rare and diagnostic biopsies are relied on for genetic information. This can be problematic because of tumor heterogeneity and tumor evolution. Firstly, biopsies only sample one portion of the tumor, and because tumors are heterogeneous, this will cover the full genetic landscape of the tumor. Secondly, after treatment tumors evolve and there may be new mutations not represented in the diagnostic sample.
Biopsy-free tumor genotyping, by way of CAPP-Seq and ctDNA, addresses many of these issues. A simple blood test is non-invasive and much safer and easier to subject cancer patients to multiple times through the course of treatment. Using ctDNA gives a better sample of tumor DNA compared to a single area of a tumor collected in a biopsy, allowing a better estimate of tumor heterogeneity. Taking multiple samples of ctDNA at different time points following the course of treatment allows tumor evolution to be uncovered. This can help detect the emergence of mutations that confer resistance to a targeted therapy and allow the course of treatment to be adjusted accordingly. CAPP-Seq specifically allows for the screening of multiple genomic locations which will become important as the list of cancer mutations important for treatment continues to grow. In a study for late stage NSCLC, they performed a version of CAPP-Seq where the tumor biopsy was not sequenced first, and they were able to correctly classify 100% of patient plasma samples with a 0% false positive rate. This shows that even without previous knowledge of tumor mutations, they can be accurately discovered by ctDNA alone.