
Development status Active Operating system License GPL | ||
 | ||
Stable release 1.5.0 / 1 July 2015; 19 months ago (2015-07-01) | ||
BioRuby is a collection of Open Source Ruby code, comprising classes for computation molecular biology and bioinformatics. It contains classes for DNA and protein sequence analysis, sequence alignment, biological database parsing, structural biology and other bioinformatics tasks.
Contents
- BioRuby
- Version history
- Installation
- Mac OS XUnixLinux
- Windows
- Usage
- Basic Syntax
- String to BioSequence object
- BioSequence object to String
- Translating a DNA or RNA Sequence or SymbolList to Protein
- Translating a single codon to a single amino acid
- Writing Sequences in Fasta format
- Reading in a Fasta file
- Major Classes
- Basic Data Structure
- Databases and sequence file formats
- Wrapper and parsers for bioinformatics tool
- File network and database IO
- Biogem
- Plugins
- References
BioRuby is released under the GNU GPL version 2 or Ruby licence and is one of a number of Bio* projects, designed to reduce code duplication.
In 2011, the BioRuby project introduced the Biogem software plugin system and are listed on biogems.info, with two or three new plugins added every month.
BioRuby is managed via the bioruby.org website and BioRuby GitHub repository.
BioRuby
The BioRuby project was first started in 2000 by Toshiaki Katayama as a Ruby implementation of similar bioinformatics packages such as BioPerl and BioPython. The initial release of version 0.1 has been frequently updated by contributors both informally and at organised “hackathon” events, with the most recent release of version 1.4.3.0001 in May 2013.
In June 2005, BioRuby was funded by IPA as an Exploratory Software Project, culminating with the release of version 1.0.0 in February 2006.
BioRuby has been the focus of a number of Google Summer of Code projects, including;
Version history
The above list of releases is abbreviated; the full list can be found here.
Installation
BioRuby is able to be installed onto any instance of Ruby; as Ruby is a highly cross platform language, BioRuby is available on most modern operating systems.
It is required that Ruby be installed prior to BioRuby installation.
Mac OS X/Unix/Linux
Mac OS X has Ruby and RubyGems installed by default and for Unix/Linux installation of RubyGems is recommended.
If Ruby and RubyGems are installed, BioRuby can be installed using this command via the terminal;
If you need to install from the source code distribution, obtain the latest package from the archive and in the bioruby source directory, run the following commands;
Windows
Installation via RubyGems is highly recommended; this requires Ruby and RubyGems be installed, then the following command run at the command prompt;
Usage
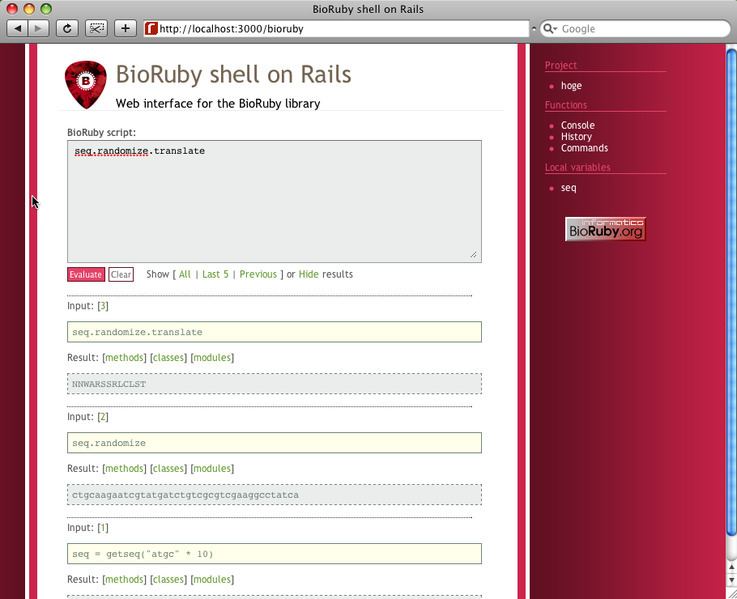
BioRuby can be accessed via the terminal, Ruby IDEs or via a BioRubyOnRails implementation. Instructions for the installation and use of BioRubyOnRails can be found at bioruby.open-bio.org/wiki/BioRubyOnRails.
Basic Syntax
The following are examples of basic sequence manipulations using BioRuby. You can find more syntax examples at bioruby.open-bio.org/wiki/SampleCodes#.
String to Bio::Sequence object
Parsing a string into Bio::Sequence object.
Bio::Sequence object to String
This an example that showcases the simplicity of BioRuby. It does not require any method call to convert the sequence object to a string.
Parsing a sequence object into a string.
Translating a DNA or RNA Sequence or SymbolList to Protein
There is no need to convert DNA sequence to RNA sequence or vice versa before its translation in BioRuby. You can simply call a translate method for Bio::Sequence::NA object.
Translating a single codon to a single amino acid
The general translation example shows how to use the translate method of Bio::Sequence::NA object but most of what goes on is hidden behind the convenience method. If you only want to translate a single codon into a single amino acid you get exposed to a bit more of the gory detail but you also get a chance to figure out more of what is going on under the hood.
Another way to do this is the following
Writing Sequences in Fasta format
To print out any Bio::Sequence object in FASTA format, All you need is to call is "puts objectName.is_fasta()"
Reading in a Fasta file
This program opens FASTA format file for reading and iterates on each sequence in the file.
This program automatically detects and reads FASTA format files given as its arguments.
Similar but specify FASTA format explicitly.
See more syntax examples on SampleCodes
Major Classes
The below classes have been identified by a group of major code contributors as major classes.
Basic Data Structure
These classes allow you to natively store complicated biological data structure effectively.
Databases and sequence file formats
Accesses online biological databases and reads from common file-formats.
Wrapper and parsers for bioinformatics tool
These classes allow for easy access to commonly used bioinformatics tools.
File, network and database I/O
A full list of classes and modules can be found at bioruby.org/rdoc/.
Biogem
Biogem provides a set of tools for bioinformaticians who want to code an application or library that uses or extends BioRuby's core library, as well as share the code as a gem on rubygems.org. Any gem published via the Biogem framework is also listed at biogems.info.
The aim of Biogem is to promote a modular approach to the BioRuby package and simplify the creation of modules by automating process of setting up directory/file scaffolding, a git repository and releasing online package databases.
Biogem makes use of github.com and rubygems.org and requires the setup of unique accounts on these websites.
Plugins
BioRuby will have a completed plugin system at the 1.5 release.