
EC number 3.2.1.31 IntEnz IntEnz view ExPASy NiceZyme view | CAS number 9001-45-0 BRENDA BRENDA entry KEGG KEGG entry | |
 | ||
Beta-glucuronidases are members of the glycosidase family of enzymes that catalyze breakdown of complex carbohydrates. Human β-glucuronidase is a type of glucuronidase (a member of glycosidase Family 2) that catalyzes hydrolysis of β-D-glucuronic acid residues from the non-reducing end of mucopolysaccharides (also referred to as glycosaminoglycans) such as heparan sulfate. Human β-glucuronidase is located in the lysosome. In the gut, brush border β-glucuronidase converts conjugated bilirubin to the unconjugated form for reabsorption. Beta-glucuronidase is also present in breast milk, which contributes to neonatal jaundice. The protein is encoded by the GUSB gene.
Contents
Structure
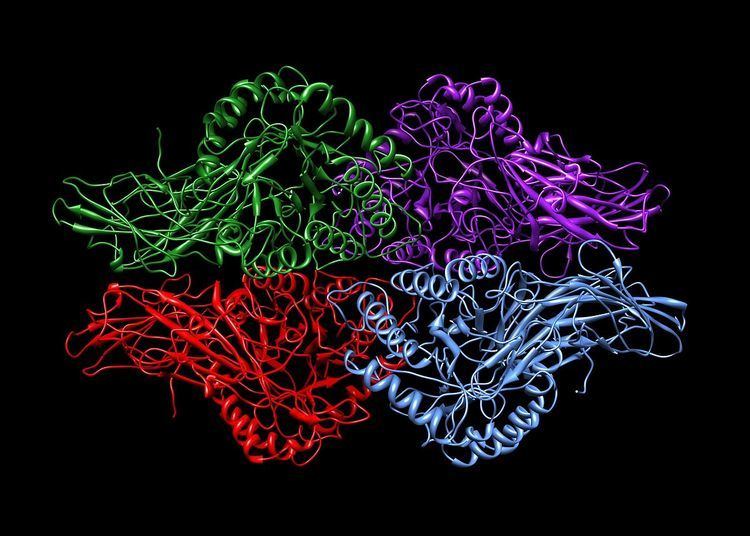
Human β-glucuronidase is synthesized as an 80 kDa monomer (653 amino acids) before proteolysis removes 18 amino acids from the C-terminal end to form a 78 kDa monomer. Beta-glucuronidase exists as a 332 kDa homotetramer. Beta-glucuronidase contains several notable structural formations, including a type of beta barrel known as a jelly roll barrel and a TIM barrel.
Mechanism of catalysis
Human β-glucuronidase is homologous to the Escherichia coli enzyme β-galactosidase. This homologous relationship, along with the knowledge that glycosidases often perform hydrolysis catalyzed by two acidic residues, enabled the development of a mechanistic hypothesis. This hypothesis proposes that the two glutamic acid residues Glu540 and Glu451 are the nucleophilic and acidic residues, respectively, and that the tyrosine residue Tyr504 is also involved in catalysis. In support of this hypothesis, experimental mutations in any of these three residues result in large decreases of enzymatic activity. Increased activity of an E451A mutant enzyme (where Glu451 is replaced with an alanine residue) after addition of azide is consistent with Glu451 as the acid/base residue. Using analysis of labeled β-glucuronidase peptides after hydrolysis of a substrate that enters a very stable intermediate stage, researchers have determined that Glu540 is the nucleophilic residue.
Though the particular type of nucleophilic substitution employed by β-glucuronidase is unclear, evidence for the mechanisms of their homologues in the glycosidase family suggests that these reactions are qualitatively SN2 reactions. The reactions proceed through a transition state with oxocarbenium ion characteristics. Initially, these mechanisms, because of this oxocarbenium characteristic of the transition state, were suggested to be SN1 reactions proceeding through a discrete oxocarbenium ion intermediate. However, more recent evidence suggests that these oxocarbenium ion states have lifetimes of 10 femtoseconds - 0.1 nanoseconds (similar to that of a bond vibration period). These lifetimes are too short to assign to a reaction intermediate. From this evidence, it appears that these reactions, while having an SN1 appearance due to the oxocarbenium ion characteristics of their transition states, must be qualitatively SN2 reactions.
The specific activity of Tyr504 in the catalytic mechanism is unclear. Through comparison to the structural data of the homologous enzyme xylanase, it has been suggested that Tyr504 of β-glucuronidase might stabilize the leaving nucleophile (Glu540) or modulate its activity.
In addition to these residues, a conserved asparagine residue (Asn450) has been suggested to stabilize the substrate through the action of a hydrogen bond at the 2-hydroxyl group of the sugar substrate.
Sly syndrome
Deficiencies in β-glucuronidase result in the non recessive inherited metabolic disease known as Sly syndrome or Mucopolysaccharidosis VII. A deficiency in this enzyme results in the build-up of non-hydrolyzed mucopolysaccharides in the patient. This disease can be extremely debilitating for the patient or can result in hydrops fetalis prior to birth. In addition, mental retardation, short stature, coarse facial features, spinal abnormalities, and enlargement of liver and spleen are observed in surviving patients. This disease has been modeled in a strain of mice as well as a family of dogs. More recently researchers have discovered a feline family that exhibits deficiencies in β-glucuronidase activity. The source of this reduction of activity has been identified as an E351K mutation (Glu351 is mutated to a lysine residue). Glu351 is conserved in mammalian species, which suggests an important function for this residue. Examination of the human X-ray crystal structure suggests that this residue (Glu352 in the human enzyme), which is buried deep within the TIM barrel domain, may be important for stabilization of the tertiary structure of the enzyme. In the crystal structure, it appears that Arg216, a member of the jelly roll domain of the protein, forms a salt bridge with Glu352; therefore, Glu352 is likely involved in stabilizing the interaction between two different three-dimensional domains of the enzyme.
Molecular applications: use as a reporter gene
In molecular biology, β-glucuronidase is used as a reporter gene to monitor gene expression in mammalian and plant cells. Monitoring β-glucuronidase activity through the use of a GUS assay allows determination of the spatial and temporal expression of the gene in question.