
ICD-9-CM 743.44 | DiseasesDB 30800 | |
 | ||
Axenfeld syndrome (also known as Axenfeld-Rieger syndrome or Hagedoom syndrome) is a rare autosomal dominant disorder, which affects the development of the teeth, eyes, and abdominal region.
Contents
Eponym
It is named after the German ophthalmologist Theodor Axenfeld who studied anterior segment disorders, especially those such as Rieger Syndrome and the Axenfeld Anomaly.
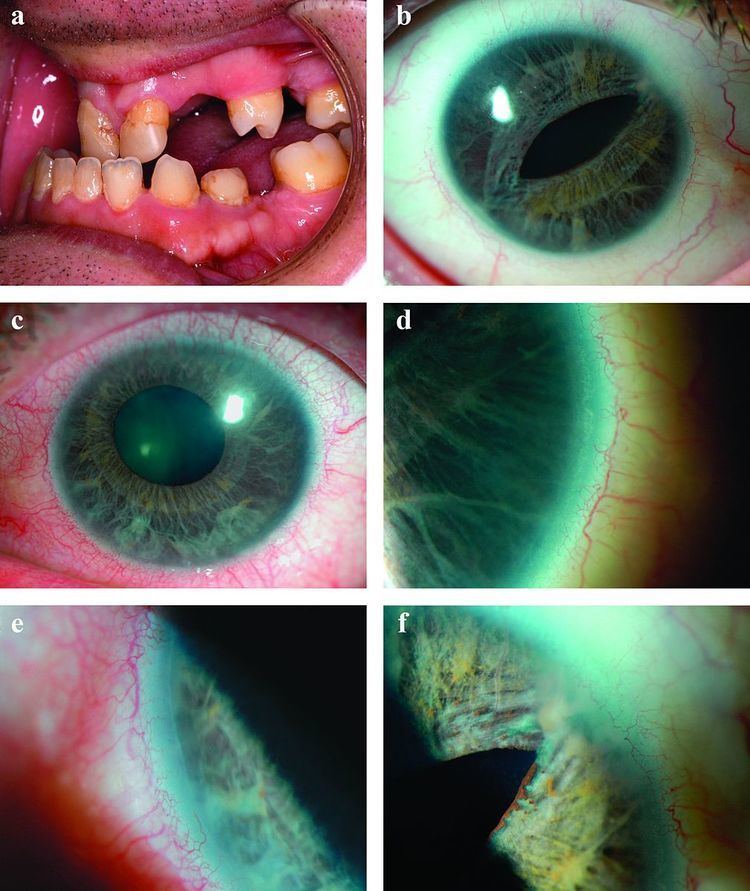
Axenfeld-Rieger syndrome is characterized by abnormalities of the eyes, teeth, and facial structure. Rieger Syndrome, by medical definition, is determined by the presence of malformed teeth, underdeveloped anterior segment of the eyes, and cardiac problems associated with the Axenfeld anomaly. The term "Rieger syndrome" is sometimes used to indicate an association with glaucoma. Glaucoma occurs in up to 50% of patients with Rieger Syndrome. Glaucoma develops during adolescence or late-childhood, but often occurs in infancy. In addition, a prominent Schwalbe's line, an opaque ring around the cornea known as posterior embryotoxon, may arise with hypoplasia of the iris. Below average height and stature, stunted development of the mid-facial features and mental deficiencies may also be observed in patients.
Diagnosis
Although most recognized for its correlation with the onset of glaucoma, the malformation is not limited to the eye, as Axenfeld syndrome when associated with the PITX2 genetic mutation usually presents congenital malformations of the face, teeth, and skeletal system.
The most characteristic feature affecting the eye is a distinct corneal posterior arcuate ring, known as an "embryotoxon". The iris is commonly adherent to the Schwalbe's line (posterior surface of the cornea).
Diagnosis One of the three known genetic mutations which cause Rieger Syndrome can be identified through genetic samples analysis. About 40% of Axenfeld-Rieger sufferers display mutations in one of the three genes known as PAX6,[8][9] PITX2 and FOXC1. The difference between Type 1, 2, and 3 Axenfeld Syndrome is the genetic cause, all three types display the same symptoms and abnormalities.
The OMIM classification is as follows:
Detection of any of these mutations can give patients a clear diagnosis and prenatal procedures such as preimplantation genetic diagnosis, Chorionic villus sampling and Amniocentesis can be offered to patients and prospective parents.
Pathophysiology
The molecular genetics of Axenfeld syndrome are poorly understood, but centers on three genes identified by cloning of chromosomal breakpoints from patients.
This disorder is inheritable as an autosomal dominant trait, which means the defective gene is located on an autosome, and only one copy of the gene is sufficient to cause the disorder when inherited from a parent who has the disorder. As shown in the diagram, this gives a 50/50 chance of offspring inheriting the condition from an affected parent.