
 | ||
The asymmetric Keck allylation is a chemical reaction that involves the addition of allyl group to an aldehyde.
Contents
Background
Laszlo Kuerti and Barbara Czako provide a superb review of many reactions in organic chemistry, including the Keck allylation; their text describes the importance of this reaction, the themes of which have been ordered similarly here. Secondary homoallylic alcohols are important moieties in organic synthesis, used in intermediates in the production of (−)-Gloeosporone, Epothilone A, the CD-Subunit of spongistatins, and the C10-C20 Subunit of rhizoxin A, among other natural products. The Keck allylation has also been utilized to form substituted tetrahydropyrans enantioselectively, moieties found in products such as phorboxazole and bryostatin 1. The general reaction scheme follows:
Although the groups of E. Tagliavini and K. Mikami reported the catalysis of this reaction using a Ti(IV) BINOL complex in the same year,Keck’s seminal publication reported asymmetric allylation with higher enantio- and diastereoselectivity using Ti(IV)-BINOL prepared by mixing Ti(Oi-Pr)4 in dichloromethane at room temperature for between 5 minutes and 1 hour, without the use of 4 Angstrom molecular sieves as in Mikami’s procedure, or excess of BINOL as in Tagliavini’s procedure. Keck’s early success with stereoselectivity and the simplicity of the catalyst preparation led to many improvements in reaction design, including development of BINOL derivatives, addition of stoichiometric additives to enhance rate, and extension of scope to substituted stannane nucleophiles.
Mechanism
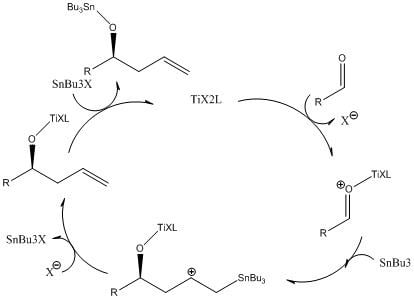
The mechanism of this allylation is not fully known, although a cycle involving activation of the aldehyde by the bimeric BINOL-Ti complex followed by the addition of the allyl ligand to the aldehyde, removal of the tributyltin, and transmetallation to regenerate the Ti complex has been proposed.
Work performed by Keck and followed up by Faller and coworkers showed a positive nonlinear effect (NLE) correlating the product enantiomeric purity with the BINOL enantiomeric purity. These observations imply that a dimeric meso-chiral catalyst is less active than the homochiral dimers, leading to the observed chiral amplification.
Corey and coworkers established a hydrogen bonding model that accounts for the absolute stereochemistry of the transformation.
Improvements
The Tagliavini group, which had carried out asymmetric allylation using a similar BINOL-Ti(IV) complex, followed up early successes by synthesizing a variety of enantiopure substituted binaphthyl ligands. The most successful of these substituted binaphthyls, shown below, gave 92% product enantiomeric excess in the addition of allyltributyltin to aldehydes with a Ti(OiPr)2Cl2 metal complex.
The Brenna group developed a synthesis for a binol analog, shown below, which can be resolved into its enantiomers quite easily and used as a chiral auxiliary for stereoselective Keck allylations, showing in some cases improved enantiomeric excesses of up to 4% over the (R)-BINOL catalyzed allylations. Additionally, the developed auxiliary also showed an NLE similar to the classic BINOL, allowing enantio-impure quantities to be used.
Faller’s group, whose aforementioned work helped elucidate the chiral amplification of the reaction, also developed the use of diisopropyl tartrate in a chiral poisoning strategy. Diisopropyl tartrate, racemic BINOL, Ti(OiPr)4, phenylaldehyde, and allyltributyltin were used to give enantiomeric excesses of up to 91%. Yoshida and coworkers developed a synthesis of dendritic binaphthols that serve as homogenous, easily recoverable catalyst systems, and showed that they were amenable to forming homoallylic alcohols using Keck’s allylation conditions. The general structural outline of these species is shown below.
Maruoka and Kii developed a bidentate Ti(IV) binol ligand for the allylation of aldehydes with the aim of restricting M-O bond rotation between the lewis acid and the aldehyde in order to improve enantiomeric excesses. The bidentate ligand, shown below, containing two titaniums, binols, and an aromatic diamine connecting moiety, gave enantiomeric excesses of up to 99%. Improved stereoselectivity is proposed to come from double activation of the carbonyl from the titaniums, a hypothesis supported by C13 NMR and IR spectroscopy studies on 2,6-γ-pyrone substrate. The most convincing evidence that the M-O rotation is restricted comes from NOE NMR studies on trans-4-methoxy-3-buten-2-one. Radiation of methoxyvinyl protons in free enone and in enone complexed with monodentate Ti(IV) show s-cis and s-trans conformations, while radiation of the enone in a bidentate Ti(IV) complex showed predominantly s-trans conformers. In 2003, this group extended the allylation strategy using this bidentate catalyst to ketones.
Two key steps in the allylation reaction involve breakage of the Sn-C bond in the allyl fragment and formation of the O-Sn bond to facilitate reproduction of the Ti(IV) catalyst. Chan Mo-Yu and coworkers developed an alkylthiosilane accelerator to promote both of these steps, simultaneously increasing the reaction rate and lowering the required catalyst dosages. Coupling of phenylaldehyde with allyltributyltin afforded 91% yield and 97% enantiomeric excess of homoallylic alcohol using 10mol% of the BINOL-Ti(IV) catalyst, however addition of the alkylthiosilane and use of only 5mol% catalyst gave 80% yield and 95% enantiomeric excess of homoallylic alcohol. The scheme involving the alkylthiosilane is shown below.
Brueckner and Weigand extended the use of this allylation chemistry to beta-substituted stannanes, including those that contain heterocycles, in 1996, exploring a variety of titanium alkoxides, premixing times, and reaction temperatures. The optimal discovered conditions were 10 mol% Ti(OiPr)4 or Ti(OEt)4, 20mol% enantiopure BINOL, with a premixing period of 2 hours, giving enantiomeric excesses of up to 99%. The general reaction scheme is shown below.