
Specialty medical genetics ICD-9-CM 743.45 DiseasesDB 723 | ICD-10 Q13.1 OMIM 106200 106210 eMedicine oph/43 | |
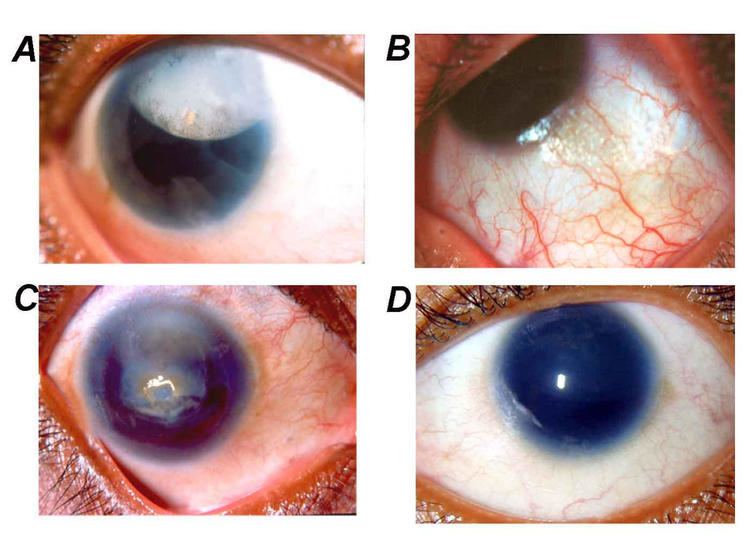 | ||
Aniridia is the absence of the iris, usually involving both eyes. It can be congenital or caused by a penetrant injury. Isolated aniridia is a congenital disorder which is not limited to a defect in iris development, but is a panocular condition with macular and optic nerve hypoplasia, cataract, and corneal changes. Vision may be severely compromised and the disorder is frequently associated with a number of ocular complications: nystagmus, amblyopia, buphthalmos, and cataract. Aniridia in some individuals occurs as part of a syndrome, such as WAGR syndrome (kidney nephroblastoma (Wilms tumour), genitourinary anomalies and intellectual disability), or Gillespie syndrome (cerebellar ataxia).
Contents
PAX6
The AN2 region of the short arm of chromosome 11 (11p13) includes the PAX6 gene (named for its PAired boX status), whose gene product helps regulate a cascade of other genetic processes involved in the development of the eye (as well as other non-ocular structures). This PAX6 gene is around 95% similar to the pax gene found in zebrafish, a creature whose ancestors diverged from human evolutionary development around 400 million years ago. Thus the PAX6 gene is highly conserved across evolutionary lineages.
Defects in the PAX6 gene cause aniridia-like ocular defects in mice (as well as Drosophila). Aniridia is a heterozygous disorder, meaning that only one of the two chromosome 11 copies is affected. When both copies are altered (homozygous condition), the result is a uniformly fatal condition with near complete failure of entire eye formation. In 2001, two cases of homozygous aniridia patients were reported; the fetuses died prior to birth and had severe brain damage. In mice, homozygous small eye defect (mouse Pax-6) leads to loss of the eyes and nose and the murine fetuses suffer severe brain damage.
Types
Aniridia may be broadly divided into hereditary and sporadic forms. Hereditary aniridia is usually transmitted in an autosomal dominant manner (each offspring has a 50% chance of being affected), although rare autosomal recessive forms (such as Gillespie syndrome) have also been reported. Sporadic aniridia mutations may affect the WT1 region adjacent to the AN2 aniridia region, causing a kidney cancer called nephroblastoma (Wilms tumor). These patients often also have genitourinary abnormalities and intellectual disability (WAGR syndrome).
Several different mutations may affect the PAX6 gene. Some mutations appear to inhibit gene function more than others, with subsequent variability in the severity of the disease. Thus, some aniridic individuals are only missing a relatively small amount of iris, do not have foveal hypoplasia, and retain relatively normal vision. Presumably, the genetic defect in these individuals causes less "heterozygous insufficiency," meaning they retain enough gene function to yield a milder phenotype.
Mutational Analysis
Molecular (DNA) testing for PAX6 gene mutations (by sequencing of the entire coding region and deletion/duplication analysis) is available for isolated aniridia and the Gillespie syndrome. For the WAGR syndrome, high-resolution cytogenetic analysis and fluorescence in situ hybridization (FISH) can be utilized to identify deletions within chromosome band 11p13, where both the PAX6 and WT1 genes are located.