
Entrez 275 | Ensembl ENSG00000145020 | |
 | ||
Aliases AMT, aminomethyltransferase, GCE, GCST, GCVT, NKH External IDs MGI: 3646700 HomoloGene: 409 GeneCards: AMT | ||
Aminomethyltransferase is an enzyme that catabolizes the creation of methylenetetrahydrofolate. It is part of the glycine decarboxylase complex.
Contents
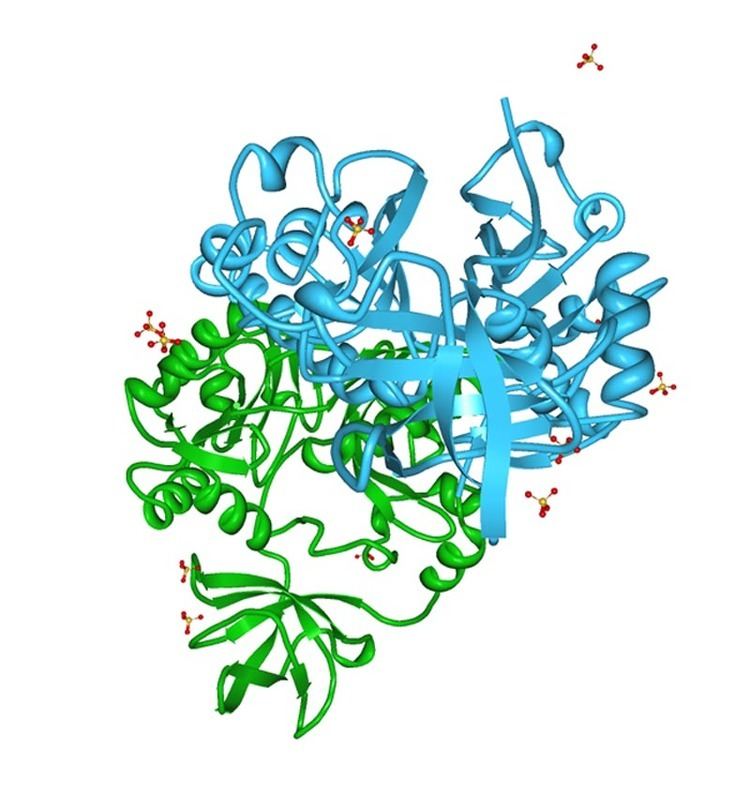
Structure
The gene is about 6 kb in length and consists of nine exons. The 5′-flanking region of the gene lacks typical TATAA sequence but has a single defined transcription initiation site detected by the primer extension method. Two putative glucocorticoid-responsive elements and a putative thyroid hormone-responsive element are present. The AMT gene has been localized to 3p21.2-p21.1 by fluorescence in situ hybridization. The 1209 base pair open reading frame encodes 403 amino acid precursor protein, and the deduced amino acid sequence of the mature peptide shows 90 and 68% homology to that of bovine and chicken counterpart, respectively.
The protein encoded by this gene has its crystal structure resolved at 2 Angstroms. The most recent model contains two monomers related by a non-crystallographic 2-fold axis, 1176 water molecules, and 11 molecules sulfate ions in an asymmetric unit. Several dimeric interactions are observed among the residues on the N-terminal loop, on α-helix D, and the flank on either side of β-strand 8 of the two monomers.
Function
The protein encoded by AMT catalyzes the release of ammonia and the transfer of a methylene carbon unit to a tetrahydrofolate moiety. The aminomethyl intermediate is the product of the decarboxylation of glycine catalyzed by P-protein. In the reverse reaction, T-protein catalyzes the formation of the H-protein-bound aminomethyl lipoate intermediate from 5,10-CH2-H4folate, ammonia, and reduced H-protein via an ordered Ter Bi mechanism, in which reduced H-protein is the first substrate to bind followed by 5,10-CH2-H4folate and ammonia.
Clinical significance
Mutations in the AMT gene are associated with Glycine encephalopathy, also known as nonketotic hyperglycinemia (NKH), which is an inborn error of glycine metabolism defined by deficient activity of the glycine cleavage enzyme and, as a consequence, accumulation of large quantities of glycine in all body tissues including the brain. The majority of glycine encephalopathy presents in the neonatal period (85% as the neonatal severe form and 15% as the neonatal attenuated form). Of those presenting in infancy, 50% have the infantile attenuated form and 50% have the infantile severe form. Overall, 20% of all children presenting as either neonates or infants have a less severe outcome, defined as developmental quotient greater than 20. A minority of patients have mild or atypical forms of glycine encephalopathy. The neonatal form manifests in the first hours to days of life with progressive lethargy, hypotonia, and myoclonic jerks leading to apnea and often death. Surviving infants have profound intellectual disability and intractable seizures. The infantile form is characterized by hypotonia, developmental delay, and seizures. The atypical forms range from milder disease, with onset from late infancy to adulthood, to rapidly progressing and severe disease with late onset. Glycine encephalopathy is suspected in individuals with elevated glycine concentration in blood and CSF. An increase in CSF glycine concentration together with an increased CSF-to-plasma glycine ratio suggests the diagnosis. Enzymatic confirmation of the diagnosis relies on measurement of glycine cleavage system (GCS) enzyme activity in liver obtained by open biopsy or autopsy. The majority of affected individuals have no detectable enzyme activity. The three genes in which biallelic mutations are known to cause glycine encephalopathy are: GLDC (encoding the P-protein component of the GCS complex and accounting for 70%-75% of disease), AMT (accounting for ~20% of disease), and GCSH (encoding the H-protein component of the GCS complex and accounting for <1% of disease). About 5% of individuals with enzyme-proven glycine encephalopathy do not have a mutation in any of these three genes and have a variant form of glycine encephalopathy.