
 | ||
Aminolevulinic acid dehydratase deficiency porphyria (also known as "Doss porphyria," and "Plumboporphyria") is a neuropsychiatric condition, disease can present during early childhood (as well as in adulthood) with acute neurologic symptoms that resemble those encountered in acute intermittent porphyria. The condition is extremely rare, with fewer than 10 cases ever reported.
ALA dehydratase deficiency is a rare cause of hepatic porphyria. It is an autosomal recessive disorder that results from inappropriately low levels of the enzyme ALA dehydratase (ALAD, also called porphobilinogen synthase), which is required for normal heme synthesis.
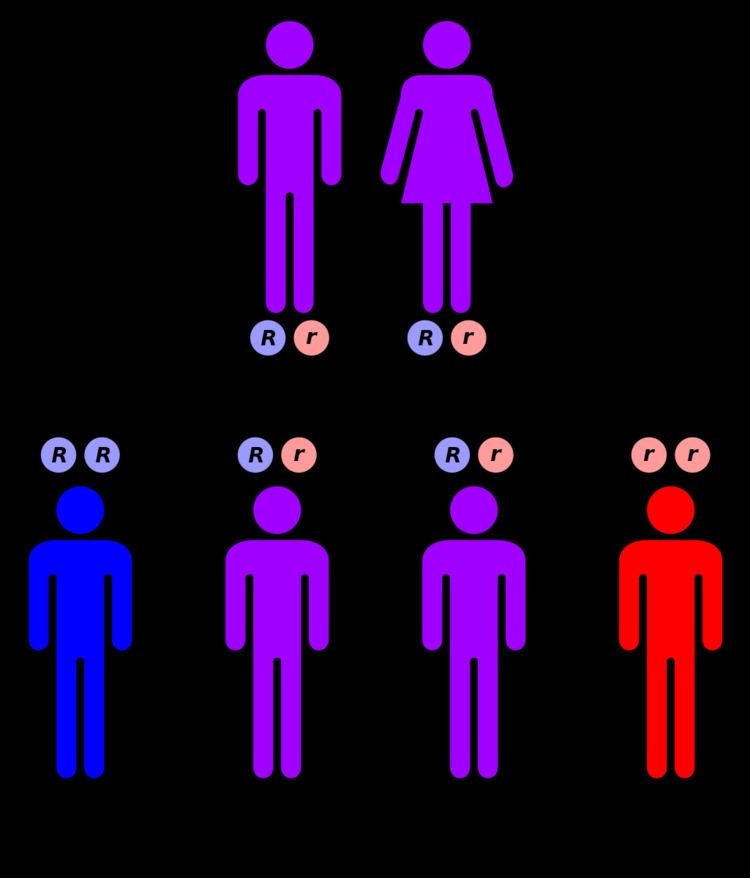
Genetics
ALA dehydratase deficiency is inherited in an autosomal recessive manner. This means a defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.