
Specialty hematology ICD-9-CM 282.43 eMedicine article/955496 | ICD-10 D56.0 DiseasesDB 448 33334 | |
 | ||
OMIM 141800 141850 142310 604131 | ||
Alpha-thalassemia (α-thalassemia, α-thalassaemia) is a form of thalassemia involving the genes HBA1 and HBA2. Alpha-thalassemia is due to impaired production of alpha chains from 1,2,3, or all 4 of the alpha globin genes, leading to a relative excess of beta globin chains. The degree of impairment is based on which clinical phenotype is present (how many genes are affected).
Contents
Signs/symptoms
The presentation of individuals with alpha-thalassemia consist of the following:
Cause
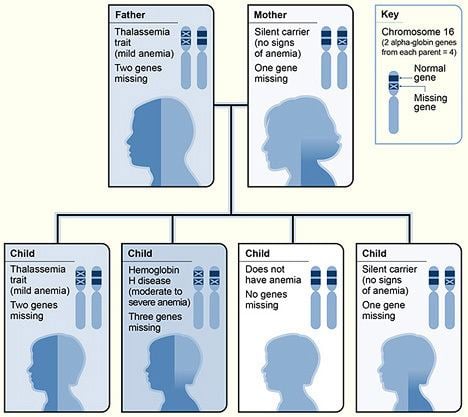
Alpha thalassemias are most commonly inherited in a Mendelian recessive manner. They are also associated with deletions of chromosome 16p. Alpha thalassemia can also be acquired under rare circumstances.
Pathophysiology
The mechanism sees that α thalassemias results in decreased alpha-globin production, therefore fewer alpha-globin chains are produced, resulting in an excess of beta chains in adults and excess γ chains in newborns. The excess β chains form unstable tetramers called Hemoglobin H or HbH of 4 beta chains. The excess γ chains form tetramers which are poor carriers of O2 since their affinity for O2 is too high so it is not dissociated in the periphery. Homozygote α0 thalassaemias, where there are lots of γ4 but no α-globins at all (referred to as Hb Barts), often result in death soon after birth.
Diagnosis
Diagnosis of alpha thalassemia is primarily via laboratory evaluation and haemoglobin electrophoresis. Alpha-thalassemia can be mistaken for iron deficiency anaemia on a full blood count or blood film as both conditions have a microcytic anaemia. Serum iron and serum ferritin can be used to exclude iron deficiency anaemia.
Types
There are two genetic loci for α globin, and thus four genes in diploid cells. Two genes are maternal in origin and two genes are paternal in origin. The severity of the α thalassemias is correlated with the number of affected α globin genes: the greater, the more severe will be the manifestations of the disease.When noting the genotype, a "α" indicates a functional alpha chain.
Treatment
Treatment for alpha thalassemia may consist of blood transfusions,and possible splenectomy, additionally gallstones may be a problem that would require surgery. Secondary complications from febrile episode should be monitored, having indicated this most individuals live without any need for treatment
Additionally, stem cell transplantation should be considered as a treatment (and cure) which is best done in early age. Other options such as gene therapy are still being developed.
Epidemiology
In terms of epidemiology, worldwide distribution of inherited alpha-thalassemia corresponds to areas of malaria exposure, suggesting a protective role. Thus, alpha-thalassemia is common in sub-Saharan Africa, the Mediterranean Basin, and generally tropical (and sub tropical) regions. The epidemiology of alpha-thalassemia in the US reflects this global distribution pattern. More specifically, HbH disease is seen in South East Asia, and the Middle East, while Hb Bart Hydrops fetalis is acknowledged in the South East Asia region only.