
 | ||
The Abramov and Pudovik reactions are the related conversions of trialkyl and dialkyl phosphites (respectively) to α-hydroxy phosphonates in the presence of carbonyl compounds. Both reactions proceed by similar mechanisms involving the attack of the nucleophilic phosphorus atom on the carbonyl carbon. They are named after Russian chemists Vasily Abramov (1904–1968) and Arkady Pudovik (1916–2006).
Contents
Introduction
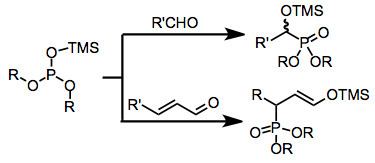
Electron-rich sources of phoshorus such as phosphites, phosphonites, and phosphinites may undergo nucleophilic addition to carbon atoms in simple carbonyl compounds. When fully esterified phosphites are used (Abramov reaction), neutralization of the resulting tetrahedral intermediate usually occurs via the transfer of an alkyl or silyl group from an oxygen attached to phosphorus to the newly created alkoxide center. Conjugate addition is also possible, and gives γ-functionalized carbonyl compounds or enol ethers after group transfer. The use of siloxy-containing phosphorus sources has greatly expanded the scope of this reaction, as the resulting α-siloxy compounds can be converted into the corresponding α-hydroxy derivatives in the presence of an alcoholic solvent
(1)
The Pudovik reaction employs dialkyl phosphites under basic conditions, which add to carbonyl compounds upon deprotonation to yield α-hydroxy phosphonates. Group transfer is not necessary in this case, as neutralization of the tetrahedral intermediate occurs via proton transfer. A catalytic, enantioselective variant of the Pudovik reaction has recently been developed.
(2)
Prevailing mechanism
Phosphites add reversibly to the carbonyl carbon of simple carbonyl compounds. Under mild conditions, reversion to the starting materials is faster than both inter- and intramolecular alkyl group transfer—the four-center transition state for intramolecular transfer exhibits poor orbital overlap. Transfer can be facilitated under conditions of high temperature or pressure. If two equivalents of aldehyde are used, addition of the tetrahedral intermediate to a second molecule of aldehyde leads either to cyclic phosphoranes 1 or linear alkyl transfer products 2.
More practical is the use of silylated phosphorus sources, which undergo intramolecular silyl group transfer in a frontside fashion, providing α-siloxy phosphorus compounds 3.
(3)
Enantioselective variants
Employing imines in the reaction allows the use of a chiral auxiliary on nitrogen. In one example, use of chiral α-methylbenzylamine provided chiral, non-racemic α-aminophosphonate.
(4)
In addition to the Lewis-acid catalyzed Pudovik reaction cited above, the reaction may be carried out in the presence of chiral amine bases. Catalytic amounts of quinine, for instance, promote the enantioselective Pudovik reaction of aryl aldehydes.
(5)
Phosphorus reagents
Phosphites are commonly used to generate α-hydroxy phosphonates. In the presence of two equivalents of aldehyde cyclic phosphoranes 1 (equation 3) predominate, but these can be easily hydrolyzed to give the corresponding hydroxy phosphonates.
(6)
When phosphonous acids are employed in the presence of catalytic amounts of base, phosphine oxides can result. The sodium salts of phosphonous acids have historically worked well in this context, and bases such as sodium amide have been used. However, asymmetric induction and selective direct addition (for conjugated carbonyl compounds) can be achieved in the presence of chiral amine bases
(7)
The discovery and use of silylated phosphorus reagents in this reaction represented a significant methodological advance. Selective silyl group transfer occurs in mixed reagents, and cleavage of the resulting silicon-oxygen bonds can often be accomplished hydrolytically, providing access to α-hydroxy derivatives. Alkylation of α-siloxy products provides a convenient route to otherwise difficult to access α-alkoxy phosphorus compounds. They can function as acyl anion equivalents when deprotonated, and give ketones after elimination under basic conditions.
(8)
Carbonyl substrates
Simple ketones and aldehydes readily undergo addition of phosphites at the carbonyl carbon. In one interesting application, addition to ketenes gives products identical to the Arbuzov reaction of acid halides.
(9)
α,β-Unsaturated ketones and aldehydes also undergo the reaction. Dienyl carbonyl substrates can experience 1,6-addition, as in the example below.
(10)
Imines can also undergo the reaction, affording α-alkylamino phosphonates. Primary amines can be produced only after acidic hydrolysis of an intermediate tert-butylamine; the use of unsubstituted imines requires very harsh conditions and gives low yields.
(11)
Synthetic utility
The α-hydroxy alkylphosphonates produced by this method can be used for additional transformations. The original carbonyl carbon is acidified by its proximity to the phosphonate group. Deprotonation at this position generates a masked acyl anion, as the phosphonate functionality can be removed after the anion reacts. Phosphonate anions can undergo alkylation and olefination (the Horner-Wadsworth-Emmons reaction). When α-amino alkylphosphonates are employed in olefination, the resulting enamines can be hydrolyzed to ketones.
(12)
Addition to unsaturated carbonyl compounds and deprotonation affords homoenolate equivalents.
Comparison with other methods
Silylated phosphite reagents are some of the most efficient for the production of α-hydroxyphosphonates. However, a few other methods exist to make these compounds. For instance, the phosphate-phosphonate rearrangement gives α-hydroxyphosphonates via a three-membered cyclic intermediate.
(13)
Experimental conditions and procedures
Generally, phosphorus addition reactions are operationally simple. Solutions of reagents in polar (acetonitrile, ethanol, tert-butanol) and non-polar (benzene) solvents may be used. Acid catalysis may be needed for additions of phosphite diesters or for in situ formation of imines. Base catalysis can also be employed in the former case. Distillation is generally sufficient to isolate pure products.