
Entrez 38 | Ensembl ENSG00000075239 | |
 | ||
External IDs OMIM: 607809 MGI: 87870 HomoloGene: 6 GeneCards: ACAT1 | ||
Acetyl-CoA acetyltransferase, mitochondrial, also known as acetoacetyl-CoA thiolase, is an enzyme that in humans is encoded by the ACAT1 (Acetyl-Coenzyme A acetyltransferase 1) gene.
Contents
Acetyl-Coenzyme A acetyltransferase 1 is an acetyl-CoA C-acetyltransferase enzyme.
Structure
The gene spans approx. 27 kb and contains twelve exons interrupted by eleven introns. The region flanking the 5’ end of the gene lacks a TATA box, but contains many GC’s and also has two CAAT boxes. The gene also may have a binding site for the transcription factor Sp1, and has sequences resembling the binding sites of several other transcription factors. Additionally, there is a 101-bp DNA fragment immediately upstream from the cap site that has promoter activity.
The human ACAT1 gene produces a chimeric mRNA through trans-splicing, a process in which separate transcripts from chromosomes 1 and 7 are spliced together. The chimeric mRNA transcript uses two sections to initiate translation: AUG(1397-1399) and GGC(1274-1276). Initiation of the first codon (AUG) results in the translation of a 50-kDa ACAT1, and initiation of the other (GGC) produces another enzymatically active 56-kDa isoform respectively; the 56kDa isoform is naturally present in human cells, including human monocyte-derived macrophages.
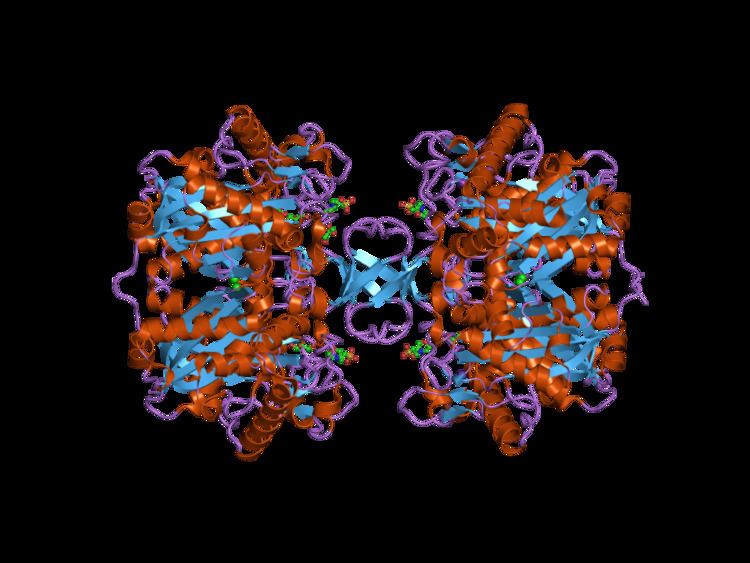
The resulting transcript encodes ACAT1, which is a 45.1 kDa protein composed of 427 amino acids. It is also a homotetrameric protein that has nine transmembrane domains (TMDs). One active residue is a Histidine at the 460th position, which is in the 7th TMD. ACAT1 has seven free Cysteine residues, but they do not affect catalytic activity. There are two functional sections of this protein, TMD7 and TMD8; one side is involved in substrate binding and catalysis, while the other is involved in subunit interactions and binding.
Function
This gene encodes a mitochondrially localized enzyme that catalyzes the reversible formation of acetoacetyl-CoA from two molecules of acetyl-CoA. The ACAT1 enzyme has a few unique properties. First, it is activated by potassium ions binding near the CoA binding site and the catalytic site. This binding causes a structural change in the active site loop. Additionally, this enzyme is able to use 2-methyl-branched acetoacetyl-CoA as a substrate, making it a unique thiolase. ACAT1 is regulated at both transcriptional and translational levels. ACAT1 enzyme activity is enhanced ACAT1’s expression is promoted transcriptionally by leptin, angiotensin II, and insulin in human monocytes/macrophages. Insulin-mediated regulation also involves ERK, p38MAPK, and JNK signaling pathways.
Ketothiolase deficiency
Mutations of the ACAT1 gene are associated with a deficiency in the encoded protein mitochondrial acetoacetyl-CoA thiolase; this is also known as ketothiolase deficiency. Many mutations have been identified in specific populations, and large scale studies have been performed to determine the allelic and genotypic frequency for the defective gene. As mitochondrial acetoacetyl-CoA thiolase is involved in beta-oxidation, a deficiency in this enzyme is marked by an increased amount of cholesterol compounds. Additionally, the isoleucine amino acid pathway is affected, such that proper metabolism of it is halted. This deficiency belongs to a more general class of disorders known as organic acidemias, in which the dysfunction of a specific step of amino acid catabolism results in the excretion of non-amino acids in the urine. This deficiency specifically presents as ketosis, acidosis, as well as hypoglycemia, but there are other clinical manifestations as well. The characteristics of organic academia disorders are vomiting, poor feeding, neurologic symptoms such as seizures and abnormal tone, and lethargy progressing to coma, which are all manifestations of toxic encephalopathy. The clinical outcome of infants with these disorders is largely determined by the time of diagnosis, with the potential outcome greatly improving if the disease is diagnosed in the first ten days of life. Ketothiolase deficiency is diagnosed by performing GC-MS and quantitative amino acid analysis in the urine; the diagnostic markers are 2-methyl-3-hydroxybutyric acid, 2-methylacetoacetic acid, and tiglylglycine. The disease is managed by trying to restore biochemical and physiologic homeostasis; common therapies include restricting diet to avoid the precursor amino acids and use of compounds to either dispose of toxic metabolites or increase enzyme activity. This disease is inherited in an autosomal recessive manner, meaning that carriers of the gene do not show symptoms of the disease.
Cancer
Additionally, expression of ACAT1 has been associated with manifestations of prostate cancer, in that ACAT1 is more significantly expressed in aggressive prostate cancer tissue samples when compared to its expression in benign cells.
Possible drug target
Over-expression of ACAT-1 is associated with more aggressive pancreatic cancer, and an ACAT-1 inhibitor avasimibe (previously developed for treatment of atherosclerosis) is being studied in cancer cell lines and an orthotopic mouse model.