
 | ||
Serotonin 5-HT2 receptors are stimulated by monoamine neurotransmitters including serotonin, dopamine and norepinephrine. 5-HT2 receptor stimulation causes a buildup of intracellular inositol triphosphate and thereby an increase of cytosolic Ca2+. 5-HT2C receptor agonists are attractive drug targets that have potential use in the treatment of a number of conditions including obesity, psychiatric disorders, sexual dysfunction and urinary incontinence.
Contents
- Discovery
- History
- Mechanism of action
- The 5 HT2C receptors and ligand binding
- Serotonin binding to 5 HT2C
- Pharmacophore
- Structure activity relationships
- Functional selectivity
- Obesity
- Lorcaserin
- Psychiatric disorders
- Vabicaserin
- Sexual dysfunction
- Urinary incontinence
- Current status
- References
The 5-HT2C receptors are one of three subtypes that belong to the serotonin 5-HT2 receptor subfamily along with 5-HT2A and 5-HT2B receptors. The development of 5-HT2C agonists has been a major obstacle, because of severe side effects due to a lack of selectivity over 5-HT2A and 5-HT2B receptors. Activation of 5-HT2A receptors can induce hallucinations, and the activation of 5-HT2B receptors has been implicated in cardiac valvular insufficiency and possibly in pulmonary hypertension.
Discovery
In the late 1960s, non-selective serotonin receptor antagonists demonstrated a relationship between serotonin receptors and food intake. Later, animal studies showed that serotonin receptor agonists might act as a mediator of satiety. Serotonin has been implicated as a critical factor in the short-term regulation of food intake and in promoting loss of weight associated with hyperphagia. Studies using pharmacological and genetic tools demonstrated that the 5-HT2C receptor subtype was one of the principal mediators through which serotonin exerts its anorectic effects in rodents. Subsequently, these receptors became a promising pharmacotherapeutic target for further investigation for the treatment of obesity. The development of 5-HT2C receptor knockout mice in the mid-1990s was a hallmark achievement in the identification and development of serotonergic drugs for weight loss. These knockout mice were hyperphagic, which led to obesity, partial Leptin resistance, increased adipose deposition, insulin resistance, and impaired glucose tolerance. As a result of these symptoms, the researchers identified a functional role for the receptors in serotonergic regulation of food intake and body weight. Later, 5-HT2C receptors were proposed as a therapeutic target for the treatment of multiple central nervous system (CNS) disorders including: psychiatric disorders, obesity, sexual dysfunction and urinary incontinence.
History
The 5-HT2c receptor agonist Fenfluramine (market names Pondimin, Ponderax and Adifax) was discovered in 1972 as a result of research performed to identify anorectic compounds lacking the effects of psycho-stimulants and sympathomimetic agents (such as amphetamines). Prior to the discovery of fenfluramine, amphetamines were the primary form of anorectic drugs available, however the side effects made them difficult to use. Fenfluramine's anorectic effect is achieved through an increase in serotonin levels, imparting a sensation of fullness, which leads to a lower intake of food. Fenfluramine was sold as a racemic mixture of two enantiomers, dexfenfluramine and levofenfluramine.
In 1994, sales of the combination drug Fen-phen (fenfluramine and phentermine) increased dramatically, as this combination produced substantial and apparent synergistic effect in promoting weight loss. Subsequently, reports of severe side effects associated with heart valve abnormalities and an increased risk of pulmonary hypertension resulted in a decision to remove products containing fenfluramine from the U.S market, and then from other markets around the world.
Dexfenfluramine inhibits serotonin reuptake, stimulating the release of serotonin. In 1996, dexfenfluramine became the first long-term treatment anti-obesity medication approved in the US; adverse effects observed during clinical trials included dry mouth, diarrhea and drowsiness. In the mid-1990s the US FDA approved dexfenfluramine as a weight loss drug. After several reports of adverse cardiovascular effects, the FDA banned dexfenfluramine in 1997.
It appears that the 5-HT2B receptors, expressed in cardiac valves, are responsible for the valvulopathies reported from the use of fenfluramine and dexfenfluramine.
The serotonin receptor agonist mCPP has a significant affinity for 5-HT2C receptors. mCPP patients experience multiple side effects due to non-selectivity over 5-HT2A and 5-HT2B receptors. The absence of the hypophagic (reduced food consumption) effect of mCPP in 5-HT2C receptor knockout mice suggests that this effect is mediated through 5-HT2C receptor activation. Repeated administration of mCPP to humans might result in decreased food intake and weight loss. mCPP is used as a prototype research tool for drug discovery of selective 5-HT2C receptor agonists.
Mechanism of action
The 5-HT2C receptors are G protein–coupled receptors that are coupled to phospholipase C (PLC) via Gαq, phospholipase A2 (PLA2), and possibly Gα13. PLC metabolizes phosphatidylinositol 4,5-bisphosphate into inositol 1,4,5-triphosphate (IP3), which regulates cellular Ca2+ flux by binding to IP3 receptors, thereby inducing the release of Ca2+. In addition, the activation of PLA2 also results in recruitment of a RhoA/PLD pathway through RhoA, an enzyme that regulates a wide spectrum of cellular functions through PLD (phospholipase D) target protein. The 5-HT2C receptors can also stimulate the extracellular signal-regulated kinase (ERK) pathway which is activated by neurotrophins and other neuroactive chemicals. Production of these chemicals effects neuronal differentiation, survival, regeneration, and structural and functional plasticity. Early studies of the ERK pathway showed that mood stabilizers for the treatment of manic-depressive illness stimulated the pathway. This led to the understanding that stimulation of the 5-HT2C receptors could regulate manic-depressive conditions in a manner similar to mood stabilizers.
5-HT2C receptors are located only within the CNS, where they can be found in several locations. The highest density of receptor expression is within the choroid plexus. Other brain locations include the nucleus of the solitary tract, dorsomedial hypothalamus, paraventricular hypothalamic nucleus and the amygdala, all of which are associated with regulation of food intake. This distribution pattern may explain the effect they have in integral function in the control of many physiological and behavioral responses, such as feeding, anxiety, temperature regulation, locomotion, sexual behavior, and the occurrence of seizures.
The 5-HT2C receptors and ligand binding
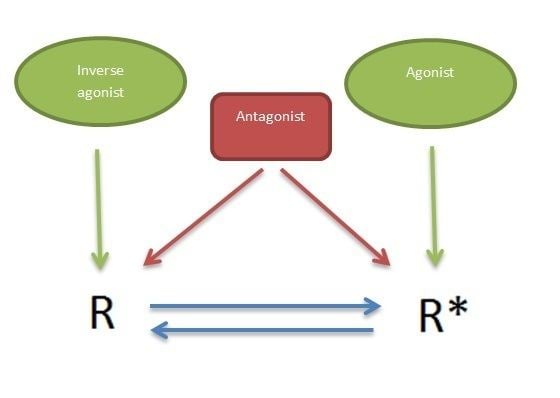
5-HT2 receptors are G protein-coupled receptors that can regulate cellular signaling in the absence of a ligand. This can be explained by a two-state model (Figure 2) where the receptor is in equilibrium between two states, an active state (R*) and an inactive state (R). Basal effector activity is defined, in part, by the absolute level of (R*), which will increase along with increasing receptor density. Ligands that preferentially bind to and stabilize the R state are termed inverse agonists and reduce the effector activity. Agonists preferentially bind to and stabilize the R* state, thereby increasing effector activity. Neutral antagonists show equal affinity for both conformations and do not alter the equilibrium between the two states, however they occupy the receptor and can block the effect of both agonists and inverse agonists.
5-HT2C and 5-HT2A receptors have a similar amino acid sequence homology, with ~50% overall sequence identity and ~80% within the TM domains, resulting in a similar pharmacological profile for the two receptors. Both receptors couple the same cellular signal transduction pathways, PLC and PLA2, that lead to an accumulation of inositol phosphate and Ca2+ within the postsynaptic cell.
The 5-HT2C receptors are the only G-protein coupled receptors known to undergo a post-transcriptional process of RNA editing. The 5-HT2C receptor gene is found on the X-chromosome, Xq24. This gene product undergoes an RNA editing process leading to a decrease in agonist binding affinity, however antagonist binding remains unaltered. This process of RNA editing generates 14 unique receptor isoforms of the 5-HT2C receptor that differ in three amino acids in the second intracellular loop.
Serotonin binding to 5-HT2C
Serotonin is an endogenous non-selective agonist for the 5-HT2C receptor with a binding constant of Ki = 16.0 nM. When serotonin binds to the receptors, the most important contacts are in TM helixes 3, 5 and 6 (Figure 3), while the other four TM helixes do not interact directly with the serotonin compound. When binding of serotonin takes place, the protonated primary amine site forms a salt bridge with D134 residue in TM 3, as well as forming a hydrogen bond with residue S138 in TM 3. The aromatic indole ring forms a strong Van der Waals interaction with residues F223 in TM 5 and F328 in TM 6. The ring falls tight into the receptor pocket, stacked between two phenylalanines. Amine of the indole group forms a hydrogen bond with S219 residue in TM 5 and hydroxide substituent of the indole forms hydrogen bonds both with residue S131 in TM 3 and I332 in TM 6. There is also a strong Van der Waals interaction between the indole and I332 in TM 6.
Pharmacophore
In the drug discovery process of a 5-HT2C agonist, a pharmacophore module has been used to discover novel 5-HT2C receptor ligands. The pharmacophore has four features; one aromatic ring, two hydrophobic features and one positive ionizable feature. Figure 4 shows an example of a compound that fits the agonist pharmacophore perfectly. The nitrogen atom of piperazine fits the positive ionizable feature, the benzofuran part fits the aromatic ring and one hydrophobic, and the trifluoromethane part fits another hydrophobic feature of the pharmacophore.
Structure-activity relationships
In a virtual screen for novel agonists, a structure-activity relationship was determined from the most potent compounds ('hits') identified. These hits contained a pyrazolo[3,4-d]pyrimidine core (shown in figure 5), which is important for potency toward the 5-HT2C receptors. Compounds with maximum potency featured two substituents linked to the core structure. The first substituent is a piperazine ring, containing a small hydrophobic group; the second substituent is a phenyl part containing a halogen- and/or oxygen-containing side chain (electronegative groups), see derivatives 1 and 2 in figure 5. Addition of aromatic groups to the piperazine ring reduces potency (derivative 4 in figure 5) and the absence of the piperazine ring or substitution with other aliphatic- or cyclic groups reduces potency as well (derivatives 5 and 6 in figure 5).
A series of 3-benzazepine derivatives, such as Lorcaserin (Figure 6) have been evaluated for their potency and selectivity for the 5-HT2C receptors. Lorcaserin is a very potent agonist, but the potency is dependent on the presence of a chloro substituent in position 8.
Arylpiperazine-containing compounds such as mCPP (Figure 7), show good potency toward the 5-HT2C receptors, but do not have sufficient selectivity for the 5-HT2C receptors over the other two receptor subtypes. Many derivatives have been examined in an attempt to increase the selectivity. Derivatives lacking the arylpiperazine core, such as 4-aryl-1,2,3,6-tetrahydropyridinum chlorine analogues, are more favorable for potency and selectivity over the other two receptors (Figure 7).
Functional selectivity
In 2016 the discovery of novel G protein biased 5-HT2C receptor agonists was published.
Obesity
Obesity is a global epidemic health problem and has received considerable attention as a major public hazard. Obesity is a chronic pathological and costly disease of abnormal or excessive fat accumulation in the body.
Studies indicate that 5-HT2C receptor activation will regulate appetite and food consumption, most likely by promoting satiety through appetite suppression by activation of 5-HT2C. Consequently, selective agents with high affinity for this receptor over 5-HT2B and 5-HT22A are being developed for the treatment of obesity.
Lorcaserin
Lorcaserin is the only agent that has completed phase III clinical trials, and achieved US Food and Drug Administration (FDA) approval. Previously approved agents were subsequently removed from the US market.
Lorcaserin is a full agonist for 5-HT2C and 5-HT2B receptors and partial agonist for 5-HT2A receptors (75% of the maximal response elicited by serotonin). Lorcaserin is a potent and selective 5-HT2C agonist with rapid oral absorption that shows dose-dependent decrease in food intake and body weight. Lorcaserin affects body weight by producing a negative energy balance through reduced food intake (energy intake) without alterations in energy expenditure and substrate oxidation. Lorcaserin has a high affinity for the 5-HT2C receptors, with 18-fold selectivity over 5-HT2A receptors and 104-fold over 5-HT2B receptors. The predicted blood concentration to stimulate 2A and 2B receptors is approximately 1400-fold for 2B and 250-fold for 2A, above the blood concentration that is required to stimulate the 2C receptors. This functional selectivity is critical to prevent potential side effects and suggests that the theoretical risk of cardiac valvulopathy is very low. Clinical trials have supported this theory since they have not revealed any side effects on heart valves or pulmonary artery pressure like the former obesity drugs. Lorcaserin is well tolerated in general, but the most frequent adverse effect are headache, nausea and dizziness.
Psychiatric disorders
Serotonin plays an important role in numerous physiological conditions. 5-HT2 receptor antagonists have long been known, but recently 5-HT2 receptor agonists are becoming promising agents in the development for new antipsychotic drugs. Historically, most pharmacological research on antipsychotic drugs have concentrated on the 5-HT2A receptor subtype. However, recent studies show that agonist activity on 5-HT2A receptors can cause hallucination. Comparison of SSRIs and the 5-HT2C receptor agonists showed that the agonists decreased immobility time and increased swimming time in the FST (forced swim test) in rats in a manner comparable to SSRIs. In the 1990s 5-HT2C receptors have received more attention as many studies have shown that selective 5-HT2C receptor agonists may be more suited in the treatment for psychotic indications.
A 5-HT2C agonist may be expected to reduce positive symptoms of schizophrenia by reducing dopamine release in the mesolimbic dopamine pathway. Vabicaserin (SCA-136) is a 5-HT2C agonist that has shown promise in preliminary testing for the treatment of schizophrenia.
Vabicaserin
Vabicaserin has a high affinity for 5-HT2C receptors and low affinity for 5-HT2B and 5-HT2A receptors. Vabicaserin is a full agonist with approximately 4-fold greater selectivity for 5-HT2C over these related receptors, in terms of binding affinity. Vabicacserin is a full agonist in stimulating the 5-HT2C receptor; it was discovered when a class of tetrahydroquinoline-fused diazepines were being researched as possible potent 5-HT2C receptor agonists.
As of 2012, Vabicaserin is in clinical trials for the treatment of schizophrenia. Long-term administration of Vabicaserin significantly decreased the number of spontaneously active mesocorticolimbic dopamine neurons without affecting nigrostriatal dopamine neurons, consistent with effects of atypical antipsychotic agents. The outcome of clinical studies for Vabicaserin may reveal whether 5-HT2C receptors can be possible targets for the treatment of schizophrenia.
Sexual dysfunction
Activation of 5-HT2C receptor subtype has been reported to mediate numerous effects, such as penile erection. Based on multiple studies, results show that several 5-HT2C receptor agonists, including mCPP and YM348 induce penile erections in rats, but mCPP seems to mimic both vasodilation and vasoconstriction. The vasodilator action is mediated by 5-HT1D receptors, whereas the vasoconstriction effect involves 5-HT2 receptor activation. YM-348 is a highly selective 5-HT2C agonist and results show that YM348 can induce penile erections and hypolocomotion (induced at a high dose) in rats, as did other 5-HT2C receptor agonists. These effects were completely inhibited by a selective 5-HT2C receptor antagonist, SB-242,084. Therefore, results suggest that YM348 is a potent and orally active 5-HT2C receptor agonist.
Urinary incontinence
Serotonin plays a key role in mechanisms involved in micturition and continence. Many potent compounds with high selectivity for 5-HT2C receptors have been synthesized and are promising candidates for further development for the treatment of stress urinary incontinence (SUI).
Current status
Many exogenous agents have been developed since the discovery of 5-HT2C receptors. Thus far a small number of agonists, with sufficient selectivity for the 5-HT2C receptors over the other subtypes have been studied in clinical trials. A variety of other 5-HT2C receptor agonists remain in pre-clinical developments, like Ro60-0175, WAY-163,909 and the inverse agonist SB-243,213. Evidence supports a therapeutic potential of 5-HT2C receptor modulation in the treatment of a variety of pathological conditions, including schizophrenia, obesity, urinary incontinence and sexual dysfunction.