
EC number 4.1.3.4 IntEnz IntEnz view ExPASy NiceZyme view | CAS number 9030-83-5 BRENDA BRENDA entry KEGG KEGG entry | |
 | ||
3-hydroxy-3-methylglutaryl-CoA lyase (or HMG-CoA lyase) is an enzyme that in human is encoded by the HMGCL gene located on chromosome 1. It is a key enzyme in ketogenesis (ketone body formation).
Contents
Structure
The HMGCL gene encodes a 34.5-kDa protein that is localized to the mitochondrion and peroxisome. Multible isoforms of the proteins are known due to alternative splicing. The major isoform (isoform 1) is most highly expressed in the liver whereas isoform 2 is found in energy-demanding tissues including the brain, heart, and skeletal muscle.
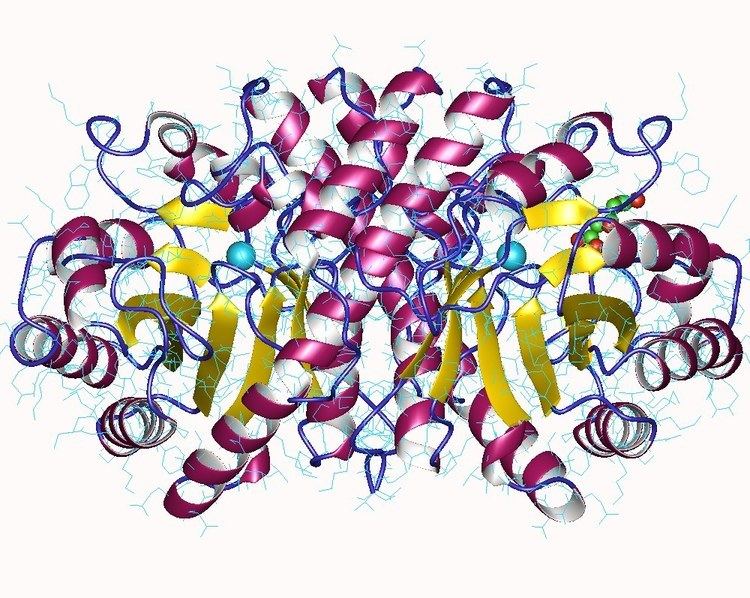
Structure of the HMGCL protein has been resolved by X-ray crystallography at 2.1-Å resolution, and reveals that the protein may function as a dimer. Substrate access to the active site of the HMGCL enzyme involves substrate binding across a cavity located at the C-terminal end of a beta barrel structure. In addition, the lysine 48 residue which is mutated in patients with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency is also found to be necessary for substrate binding.
Function
The HMGCL protein plays an essential role in breaking down dietary proteins and fats for energy. It catalyzes the reaction:
(S)-3-hydroxy-3-methylglutaryl-CoA = acetyl-CoA + acetoacetate.
and requires a divalent metal ion as co-factor.
The enzyme is required for ketogenesis in the liver, and is also responsible for processing the amino acid leucine inside the mitochondrion
Clinical Significance
Mutations in the HMGCL gene cause 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (HMGCLD), a rare autosomal recessive inborn error of metabolism characterized by disruption of ketogenesis and L-leucine catabolism. To-date more than 30 different mutations including missense mutations of different residues have been associated with patients with HMGCLD in diverse families and ethnicities. HMGCLD typically presents in the first year of the patient's life after a fasting period. Clinical acute symptoms include vomiting, seizures, metabolic acidosis, hypoketotic hypoglycemia, and lethargy.
Interactions
HMGCL interacts with itself to form homodimers and homotetramers. It is also shown in yeast two-hybrid experiments to interact with DNAJA1.