
 | ||
The [2+2] photocycloaddition is a cycloaddition-type reaction – it generally entails the formation of new molecules by the reaction of two unsaturated molecules via two atoms from each molecule (hence "[2 + 2]"). As a photochemical reaction, the use of some of form of light (generally denoted h·ν) is required, as opposed to a thermal process.
Contents
Enone–alkene cycloadditions
A specialized version of the [2+2] cycloaddition involves enone and alkene as substrates. Although the photochemical concerted [2+2] cycloaddition is allowed, the reaction between enones and alkenes is stepwise and involves discrete diradical intermediates.
History
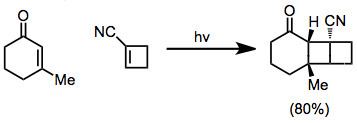
In 1908, it was discovered that exposure of carvone to "Italian sunlight" for one year gave camphor. The reaction was heavily investigated in the 1950s with emphasis on the photochemical [2+2] cycloaddition of enones to alkenes. This process can be useful for the construction of complex organic compounds. In spite of the stepwise, radical mechanism, both stereoselective intra- and intermolecular variants have emerged. Cyclic enones are employed, otherwise competitive cis-trans isomerization ensues.
Mechanism
The mechanism of [2+2] photocyclization is proposed to begin with photoexcitation of the enone to a singlet excited state. The singlet state is typically very short lived, and decays by intersystem crossing to the triplet state. At this point, the enone forms an exciplex with the ground state alkene, eventually giving the triplet diradical. Spin inversion to the singlet diradical allows closure to the cyclobutane. As an alternative a pericyclic reaction mechanism is proposed, in which after intersystem crossing a radical cation and a radical anion are formed, which then recombine to the cyclobutane.
Scope and limitations
[2+2]-Photocyclization can produce two isomers, depending on the orientation of substituents on the alkene and the enone carbonyl group. When the enone carbonyl and substituent of highest priority are proximal, the isomer is termed "head-to-head." When the enone carbonyl and substituent are distal, the isomer is called "head-to-tail." Selectivity for one of these isomers depends on both steric and electronic factors (see below).
The regiochemistry of the reaction is controlled primarily by two factors: steric interactions and electrostatic interactions between the excited enone and alkene. In their excited state, the polarity of enones is reversed so that the β carbon possesses a partial negative charge. In the transition state for the first bond formation, the alkene tends to align itself so that the negative end of its dipole points away from the β carbon of the enone.
Steric interactions encourage the placement of large substituents on opposite sides of the new cyclobutane ring.
If the enone and alkene are contained in rings of five atoms or less, double-bond configuration is preserved. However, when larger rings are used, double bond isomerization during the reaction becomes a possibility. This energy-wasting process competes with cycloaddition and is evident in reactions that yield mixtures of cis- and trans-fused products.
Diastereofacial selectivity is highly predictable in most cases. The less hindered faces of the enone and alkene react.
Intramolecular [2+2] cyclizations may give either "bent" or "straight" products depending on the reaction regioselectivity. When the tether between the enone and alkene is two atoms long, bent products predominate due to the rapid formation of five-membered rings. Longer tethers tend to give straight products.
The tether can also be attached at the 2 position of the enone. When the alkene is tethered here, bulky substituents at the 4 position of the enone enforce moderate diastereoselectivity.
A major difficulty of [2+2] photocycloadditions is the possibility of side reactions associated with the diradical intermediate or excited enone. These side reactions can often be minimized by a judicious choice of reaction conditions.
Synthetic applications
[2+2] photocyclization has been used to synthesize organic compounds with interesting topology. It is used as a key step, for instance, in a synthesis of cubane. The Favorskii rearrangement established the carbon skeleton of cubane, and further synthetic manipulations provided the desired unfunctionalized target.
Methodology
A wide variety of solvents can be used, provided they are deoxygenated to minimize reactions with oxygen. Acetone is a useful solvent, because it can serve as a triplet sensitizer. Olefin-free hexanes can be useful for intramolecular reactions. Reaction temperature can influence regioselectivity and stereoselectivity. The cycloadduct should not absorb engage in photochemistry, so the excitation wavelength is important. For intermolecular reactions, excess of the alkene should be employed to avoid competitive dimerization of the enone.
Glow sticks
Reverse [2+2] photocycloaddition, decomposition of 1,2-dioxetanedione, is stated as the mechanism that produces light in glow sticks.