
 | ||
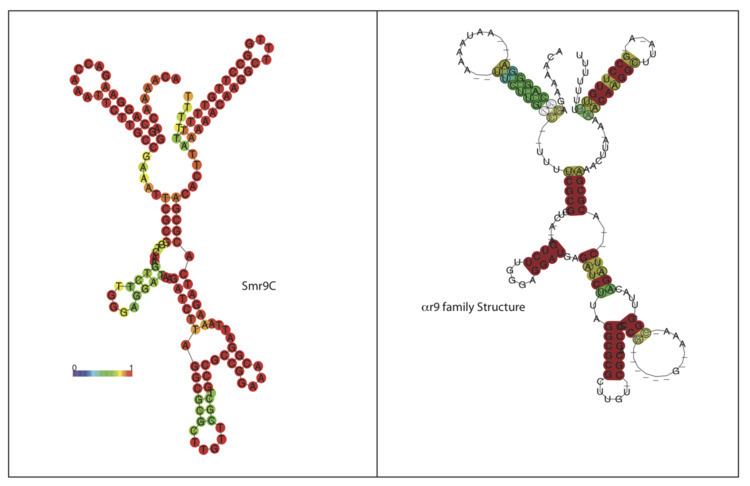
αr9 is a family of bacterial small non-coding RNAs with representatives in a broad group of α-proteobacteria from the order Rhizobiales. The first member of this family (Smr9C) was found in a Sinorhizobium meliloti 1021 locus located in the chromosome (C). Further homology and structure conservation analysis have identified full-length Smr9C homologs in several nitrogen-fixing symbiotic rhizobia (i.e. R. leguminosarum bv.viciae, R. leguminosarum bv. trifolii, R. etli, and several Mesorhizobium species), in the plant pathogens belonging to Agrobacterium species (i.e. A. tumefaciens, A. vitis, A. radiobacter, and Agrobacterium H13) as well as in a broad spectrum of Brucella species (B. ovis, B. canis, B. abortus and B. microtis, and several viobars of B. melitensis). αr9C RNA species are 144-158 nt long (Table 1) and share a well defined common secondary structure consisting of seven conserved regions (Figure 1). Most of the αr9 transcripts can be catalogued as trans-acting sRNAs expressed from well-defined promoter regions of independent transcription units within intergenic regions (IGRs) of the α-proteobacterial genomes (Figure 5).
Contents
Discovery and Structure
Smr9C sRNA was described by del Val et al. in the intergenic regions (IGRs) of the reference S. meliloti 1021 strain (http://iant.toulouse.inra.fr/bacteria/annotation/cgi/rhime.cgi). Northern hybridization experiments confirmed that the predicted smr9C locus did express a single transcript of the expected size, which accumulated differentially in free-living and endosymbiotic bacteria. TAP-based 5’-RACE experiments mapped the transcription start site (TSS) of the full-length Smr9C transcript to the 1,398,425 nt position in the S. meliloti 1021 genome (http://iant.toulouse.inra.fr/bacteria/annotation/cgi/rhime.cgi) whereas the 3’-end was assumed to be located at the 1,398,277 nt position matching the last residue of the consecutive stretch of Us of a bona fide Rho-independent terminator (Figure 5). Parallel and later studies in which Smr9C transcript is referred to as Igr#3 or sra32 independently confirmed the expression this sRNA in S. meliloti and in its closely related strain 2011. Recent deep sequencing-based characterization of the small RNA fraction (50-350 nt) of S. meliloti 2011 further confirmed the expression of Smr9C (here referred to as SmelC289), and mapped the 5’- and 3´-ends of the full-length transcript to positions 1,398,423 and 1,398,279, respectively, in the S. meliloti 1021 genome.
The nucleotide sequence of Smr9C was initially used as query to search against the Rfam database (version 10.0;
This homology search rendered no matches to known bacterial sRNA in this database. Smr9C was next BLASTed with default parameters against all the currently available bacterial genomes (1,615 sequences at 20 April 2011; http://www.ncbi.nlm.nih.gov). The regions exhibiting significant homology to the query sequence (78-89% similarity) were extracted to create a Covariance Model (CM) from a seed alignment using Infernal (version1.0) (Figure 2). This CM was used in a further search for new members of the αr9 family in the existing bacterial genomic databases.The results were manually inspected to deduce a consensus secondary structure for the family (Figure 1 and Figure 2). The consensus structure was also independently predicted with the program locARNATE with very similar predictions. The manual inspection of the sequences found with the CM using Infernal allowed finding 26 true homolog sequences, all of them present as single chromosomal copies in the α-proteobacterial genomes. The rhizobial species encoding the 12 closer homologs to Smr9C were: two R. leguminosarum trifolii strains (WSM2304 and WSM1235), two R. etli strains CFN 42 and CIAT 652, the reference R. leguminosarum bv. viciae 3841 strain, Rhizobium NGR234, and the Agrobacterium species A. vitis,A. tumefaciens, A. radiobacter and A. H13. All these sequences showed significant Infernal E-values (1.50e-39 - 14.02e-21) and bit-scores. The rest of the sequences found with the model showed high E-values between (3.40e-12 and 2.62e-04) but lower bit-scores and are encoded by Brucella species (B. ovis, B. canis, B. abortus, B. microtis, and several biobars of B. melitensis), Ochrobactrum anthropi and the Mesorhizobum species loti, M. ciceri and M. BNC.
Expression information
Parallel studies assessed Smr9C expression in S. meliloti 1021 under different biological conditions; i.e. bacterial growth in TY, minimal medium (MM) and luteolin-MM broth and endosymbiotic bacteria (i.e. mature symbiotic alfalfa nodules) and high salt stress, oxidative stress and cold and hot shock stresses. Expression of Smr9C in free-living bacteria was found to be growth-dependent, being the gene strongly down-regulated when bacteria entered the stationary phase, whereas no expression was detected in endosymbiotic bacteria. Recent deep sequencing data further revealed up-regulation of Smr9C upon salt, acidic, cold-shock and heat shock stresses. Recent co-inmuno precipitation experiments corroborate that Smr9C, does bind the bacterial protein Hfq for efficient target binding.
Promoter Analysis
All the promoter regions of the αr9 family members examined so far are very conserved in a sequence stretch extending up to 80 bp upstream of the transcription start site of the sRNA. All loci have recognizable σ70-dependent promoters showing a -35/-10 consensus motif CTTAGAC-n17-CTATAT, which has been previously shown to be widely conserved among several other genera in the α-subgroup of proteobacteria. To identify binding sites for other known transcription factors we used the fasta sequences provided by RegPredict(http://regpredict.lbl.gov/regpredict/help.html), and used those position weight matrices (PSWM) provided by RegulonDB (http://regulondb.ccg.unam.mx). We built PSWM for each transcription factor from the RegPredict sequences using the Consensus/Patser program, choosing the best final matrix for motif lengths between 14–30 bps a threshold average E-value < 10E-10 for each matrix was established, (see "Thresholded consensus" in http://gps-tools2.its.yale.edu). Moreover, we searched for conserved unknown motifs using MEME (http://meme.sdsc.edu/meme4_6_1/intro.html) and used relaxed regular expressions (i.e. pattern matching) over all Smr9C homologs promoters. This studies revealed the presence of 30 bp long region very conserved between positions -40 and -75, conserved MEME motif, (Figure 5). This sequence was present in all but one of the smrC9 homologs found, but no significant similarity to known transcription factor binding sites matrices could be established.
Genomic Context
Most of the members of the αr9 family are trans-encoded sRNAs transcribed from independent promoters in chromosomal IGRs. Exceptions are the cis-encoded antisense Smr9C homologs of A. tumefaciens and B. microti, which are located in the opposite strand of annotated genes, partially overlapping ORFs. Most of the neighboring genes of the seed alignment’s members were not annotated and thus were further manually curated. The predicted protein products of these overlapping ORFs could not be assigned to any functional category on the basis of the amino acid sequence homology. However, the genomic regions of almost all αr9 sRNAs exhibited a great degree of conservation including the sRNA-coding sequence and the upstream and downstream genes which have been predicted to code for a prolyl-tRNA syntethase (proS) and a transmembrane protein, respectively. Partial synteny of the αr9 genomic regions was observed in a few cases such as, S. medicae where instead of a proS gene an FAD-dependent pyridine nucleotide-disulfide oxidoreductase encoding gene was found upstream of the αr9 locus, and Mesorhizobium loti where no transmembrane coding gene was recognizable downstream of the sRNA gene. A special case is the Brucella group, where primary automatic annotation over their genomes identified ORFs smaller than 30 aa overlapping with the predicted αr9 sRNA in the same strand. These predicted ORFs, neither show any similarity with database entries nor any motif or signatures when searched against family and motif databases such as Interpro, PFAM or Smart, and thus, are considered here as missannotations not registered in the genomic context graph.