
 | ||
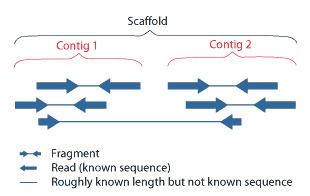
Scaffolding is a technique used in bioinformatics. It is defined as follows:
Contents
- History of Assembly Software
- Scaffolding and Next Gen Sequencing
- Celera and the Human Genome Project
- References
Link together a non-contiguous series of genomic sequences into a scaffold, consisting of sequences separated by gaps of known length. The sequences that are linked are typically contiguous sequences corresponding to read overlaps.
When creating a draft genome, individual reads of DNA are first assembled into contigs, which, by the nature of their assembly, have gaps between them. The next step is to then bridge the gaps between these contigs to create a scaffold. This can be done using either optical mapping or mate-pair sequencing.
History of Assembly Software
The sequencing of the Haemophilus Influenzae genome marked the advent of scaffolding. That project generated a total of 140 contigs, who were orientated and linked using paired end reads. The success of this strategy prompted the creation of the software, Grouper, which was included in genome assemblers. Until 2001, this was the only scaffolding software. After the HGP and Celera proved that it was possible to create a large draft genome, several other similar programs were created.
Bambus was created in 2003 and was a rewrite of the original grouper software, but afforded researchers the ability to adjust scaffolding parameters. This software also allowed for optional use of other linking data, such as contig order in a reference genome.
Scaffolding and Next Gen Sequencing
Most High-Throughput, Next Generation Sequencing (HT-NGS) platforms produce shorter read lengths compared to Sanger Sequencing. These new platforms are able to generate large quantities of data in short periods of time, but until methods were developed for de novo assembly of large genomes from short read sequences, Sanger sequencing remained the standard method of creating a reference genome. The SMRT platform is capable of generating 1000 bp reads, but the commonly used platforms of Heliscope and Illumina generate read lengths of 75 bp or less, which caused many people in the science community to doubt a reliable reference genome could ever be constructed using these technologies. The increased difficulty of contig and scaffold assembly associated with the new technologies has created a demand for powerful new computer programs and algorithms capable of making sense of the data.
For a while, algorithms for scaffolding allowed for little to no human interpretation.
Celera and the Human Genome Project
Although Celera was able save funds by neglecting to install their mate pair inserts into BACs, it became hard to scaffold the contiguous series of reads after they had been linked. For the later assembly stage, the private project had to rely on the pre-mapping accomplished by the HGP's sequence-tagged site maps, BAC maps, and clone based sequences.