
Original author(s) Operating system | Stable release RAPTOR 4.2 / Nov. 2008 | |
 | ||
Developer(s) Bioinformatics Solutions Inc. Type Protein structure prediction Website www.bioinfor.com/raptor | ||
RAPTOR is protein threading software used for protein structure prediction. It has been replaced by RaptorX, which is much more accurate than RAPTOR.
Contents
Protein threading vs. homology modeling
Researchers attempting to solve a protein's structure start their a study with little more than a protein sequence. Initial steps may include performing a PSI-BLAST or PatternHunter search to locate a similar sequences with a known structure in the Protein Data Bank (PDB). If there are highly similar sequences with known structures, there is a high probability that this protein's structure will be very similar to those known structures as well as functions. If there is no homology found, the researcher must perform either X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy, both of which require considerable time and resources to yield a structure. Where these techniques are too expensive, time-consuming or limited in scope, researchers can use protein threading software, such as RAPTOR to create a highly reliable model of the protein.
Protein threading is more effective than homology modeling, especially for proteins which have few homologs detectable by sequence alignment. The two methods both predict protein structure from a template. Given a protein sequence, protein threading first aligns (threads) the sequence to each template in a structure library by optimizing a scoring function that measures the fitness of a sequence-structure alignment. The selected best template is used to build the structure model. Unlike homology modeling, which selects template purely based on homology information (sequence alignments), the scoring function used in protein threading utilizes both homology and structure information (sequence structure alignments).
If a sequence has no significant homology found, homology modeling may not give reliable prediction in this case. Without homology information, protein threading can still use structure information to produce good prediction. Failed attempts to obtain a good template with BLAST often result in users processing results through RAPTOR.
Integer programming vs. dynamic programming
The integer programming approach to RAPTOR produces higher quality models than other protein threading methods. Most threading software use dynamic programming to optimize their scoring functions when aligning a sequence with a template. Dynamic programming is much easier to implement than integer programming; however if a scoring function has pairwise contact potential included, dynamic programming cannot globally optimize such a scoring function and instead just generates a local optimal alignment.
Pairwise contacts are very conserved in protein structure and crucial for prediction accuracy. Integer programming can globally optimize a scoring function with pairwise contact potential and produce a global optimal alignment.
Threading engines
NoCore, NPCore and IP are the three different threading engines implemented in RAPTOR. NoCore and NPCore are based on dynamic programming and faster than IP. The difference between them is that in NPCore, a template is parsed into many "core" regions. A core is a structurally conserved region. IP is RAPTOR's unique integer programming-based threading engine. It produces better alignments and models than the other two threading engines. People can always start with NoCore and NPCore. If their predictions are not good enough, IP may be a better choice. After all three methods are run, a simple consensus may help to find the best prediction.
3D structure modeling module
The default 3D structure modeling tool used in RAPTOR is OWL. Three-dimensional structure modeling involves two steps. The first step is loop modeling which models regions in the target sequence that map to nothing in the template. After all the loops are modeled and the backbone is ready, side chains are attached to the backbone and packed up. For loop modeling, a cyclic coordinate descent algorithm is used to fill the loops and avoid clashes. For side chain packing, a tree decomposition algorithm is used to pack up all the side chains and avoid any clashes. OWL is automatically called in RAPTOR to generate the 3D output.
If a researcher has MODELLER, they can also set up RAPTOR to call MODELLER automatically. RAPTOR can also generate ICM-Pro input files, with which people run ICM-Pro by themselves.
PSI-BLAST module
To make it a comprehensive tool set, PSI-BLAST is also included in RAPTOR to let people do homology modeling. People can set up all the necessary parameters by themselves. There are two steps involved in running PSI-BLAST. The first step is to generate the sequence profile. For this step, NR non-redundant database is used. The next step is to let PSI-BLAST search the target sequence against the sequences from the Protein Data Bank. Users can also specify their own database for each step.
Protein structure viewer
There are many different structure viewers. In RAPTOR, Jmol is used as the structure viewer for examining the generated prediction.
Output
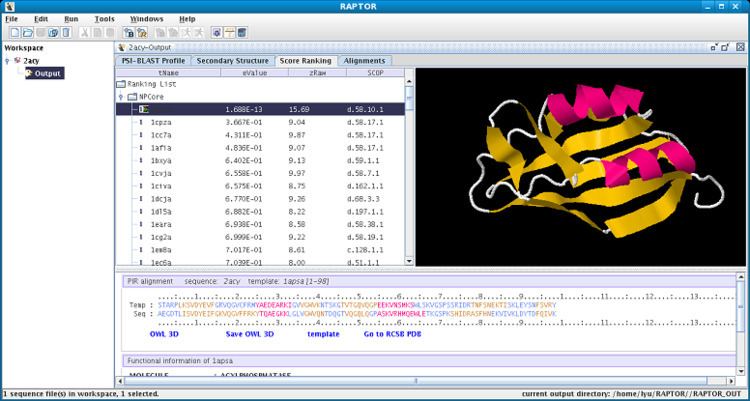
After a threading/PSI-BLAST job, one can see a ranking list of all the templates. For each template, people can view the alignment, E-value and numerous other specific scores. Also, the functional information of the template and its SCOP classification are provided. One can also view the sequence's PSM matrix and secondary structure prediction. If a template has been reported by more than one method, it will be marked with the number of times it has been reported. This helps to identify the best template.
Performance in CASP
CASP, Critical Assessment of Techniques for Protein Structure Prediction, is a biennial experiment sponsored by NIH. CASP represents the Olympic Games of the protein structure prediction community and was established in 1994.
RAPTOR first appeared in CAFASP3 (CASP5) in 2002 and was ranked number one in the individual server group for that year. Since then, RAPTOR has actively participated in every CASP for evaluation purpose and been consistently ranked in the top tier.
The most recent CASP8 ran from May 2008 until August 2008. More than 80 prediction servers and more than 100 human expert groups worldwide registered for the event, where participants attempt to predict the 3D structure from a protein sequence. According to the ranking from Zhang's group, RAPTOR ranked 2nd among all the servers (meta server and individual servers). Baker lab's ROBETTA is placed 5th in the same ranking list.