
 | ||
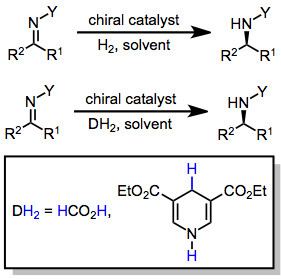
In chemistry, the hydrogenation of carbon–nitrogen double bonds is the addition of the elements of dihydrogen (H2) across a carbon–nitrogen double bond, forming amines or amine derivatives. Although a variety of general methods have been developed for the enantioselective hydrogenation of ketones, methods for the hydrogenation of carbon–nitrogen double bonds are less general. Hydrogenation of imines is complicated by both syn/anti isomerization and tautomerization to enamines, which may be hydrogenated with low enantioselectivity in the presence of a chiral catalyst. Additionally, the substituent attached to nitrogen affects both the reactivity and spatial properties of the imine, complicating the development of a general catalyst system for imine hydrogenation. Despite these challenges, methods have been developed that address particular substrate classes, such as N-aryl, N-alkyl, and endocyclic imines.
Contents
- Inner sphere mechanisms
- Outer sphere mechanisms
- Scope and limitations
- Imines
- Imine derivatives
- Applications
- Comparison with other methods
- Experimental conditions
- References
As in hydrogenation reactions of other functional groups, the reductant in C=N hydrogenations is either hydrogen gas or a transfer hydrogenation reductant such as formic acid. The process is usually catalyzed by a transition metal complex. If the complex is chiral and non-racemic and the substrate is prochiral, an excess of a single enantiomer of a chiral product can result.
Inner sphere mechanisms
The mechanism of imine hydrogenation depends on the nature of the catalyst .Catalysis by some rhodium(I) complexes proceeds through the dihydride species I. The mechanism is proposed to involve both η1 (σ-type) and η2 (π-type) coordination of the imine followed by transfer of hydrogen to generate the amine complex. Dissociation of the amine product and oxidative addition of H2 then occur, preparing the catalyst to bind another imine molecule. In some iridium-catalyzed hydrogenations, the mechanism is believed to proceed via a monohydride species. The oxidation state of iridium is always +3.
Outer sphere mechanisms
Ruthenium(II) catalysts incorporating chiral diphosphine ligands operate according to the inner-sphere mechanisms described above, in which the imine must coordinate to the catalyst and insert into the metal-hydrogen bond. Notably many ruthenium(II) catalyst operate through an "outer-sphere mechanism," during which the imine never interacts with the metal center indirectly. Instead, it receives the elements of H2 from Ru-H and N-H in a concerted, polarized fashion. This process is utilized by the Shvo catalyst and many ruthenium amine complexes.
Scope and limitations
Because the substituents attached to the imine nitrogen exert a profound influence on reactivity, few general catalyst systems exist for the enantioselective hydrogenation of imines and imine derivatives. However, catalyst systems have been developed that catalyze hydrogenation of particular classes of imines with high enantioselectivity and yield. This section describes some of these systems and is organized by the substitution pattern of the imine.
Imines
N-Aryl imines are efficiently hydrogenated using a preformed iridium(I) catalyst with low pressures of hydrogen. Although enantioselectivities vary, conversion is very high across a variety of iridium-based catalyst systems.
N-Alkyl imines are generally more difficult to reduce with high enantioselectivity than N-aryl imines. Most catalyst systems that reduce N-alkyl imines enantioselectively are based on rhodium(I) or ruthenium(II).
Endocyclic imines do not suffer from complications due to syn/anti isomerization; however, this has not proven to be a significant advantage in enantioselective hydrogenations. 3-Hydroindoles may be reduced with hydrogen gas and an iridium catalyst.
Alternatively, and illustrating the outer sphere mechanism, transfer hydrogenation from formic acid/triethylamine is catalyzed by a ruthenium(II)/diamine complex.
Heteroaromatic substrates may be reduced by hydrogen in the presence of a transition-metal complex. Although catalyst systems tend to be substrate specific, and enantioselectivities are often low, quinolines may be reduced to tetrahydroquinolies enantioselectively using several catalyst systems.
Imine derivatives
Electronically polarized N-tosyl imines are reduced with high enantioselectivity and yield by palladium(II) and ruthenium(II) catalyst systems, although catalyst loadings tend to be relatively high under both direct and transfer hydrogenative conditions.
The presence of a second coordinating group on the substrate enhances enantioselectivity in a number of cases. C-Acyl hydrazones are hydrogenated with much higher enantioselectivity than the corresponding C-alkyl hydrazones, for instance.
α-Carboxy imines are attractive precursors for α-amino acids. Organocatalytic reduction of these substrates is possible using a Hantzsch ester and a chiral phosphoric acid catalyst.
Applications
Metolachlor is the active ingredient in the widely used herbicide Dual Magnum®. A key step in its industrial production involves the enantioselective reduction of an N-aryl imine. This reduction is achieved with extremely high turnover number (albeit moderate enantioselectivity) through the use of a specialized catalyst system consisting of [Ir(COD)Cl]2, modified Josiphos ligand 3, and acid and iodide additives.
Transfer hydrogenation of endocyclic imines has been applied to the synthesis of tetrahydroisoquinoline alkaloids, such as cryspine A.
Comparison with other methods
Imines may be reduced enantioselectively using stoichiometric amounts of chiral metal hydrides. Such methods have the advantage that they are easy to implement. Reduction with hydrosilanes is a second alternative to transition-metal catalyzed hydrogenation.
Aminotransferase enzymes can be used to synthesize chiral amines in non-racemic form either through kinetic resolution of a racemate or reductive amination of a prochiral ketone. Enantioselectivities are often extremely high; however, optimization of the reaction conditions can be laborious.
Experimental conditions
The ideal conditions for imine hydrogenation depend on the nature of the substrate and catalyst system. Generally, higher pressures of hydrogen gas are needed in order to achieve reasonable reaction times; however, hydrogen pressure does not appreciably affect enantioselectivity. If the active catalyst is generated in situ, Schlenck techniques or a glove box should be employed. Parr shakers may be used for reaction pressures up to 4 bar, although precise temperature control is difficult to achieve in the reaction vessel. For higher hydrogen pressures, specialized autoclave equipment is required.
Hydrogen forms flammable mixtures with air, and any apparatus used for hydrogenation should be checked for leaks prior to the introduction of substrates and catalysts.