
Specialty medical genetics ICD-9-CM 759.89 DiseasesDB 14141 | ICD-10 Q87.3 OMIM 130650 MedlinePlus 001186 | |
Beckwith–Wiedemann syndrome (/ˈbɛkˌwɪθ ˈviːdə.mən/; abbreviated BWS) is an overgrowth disorder usually present at birth, characterized by an increased risk of childhood cancer and certain congenital features.
Contents
- Presentation
- Neoplasms
- Genetics
- Association with CDKN1C
- Management
- Prognosis
- Assisted reproductive technology
- Epidemiology
- History
- References
Common features used to define BWS are:
Presentation
Most children with BWS do not have all of these five features. In addition, some children with BWS have other findings including: nevus flammeus, prominent occiput, midface hypoplasia, hemihypertrophy, genitourinary anomalies (enlarged kidneys), cardiac anomalies, musculoskeletal abnormalities, and hearing loss. Also, some premature newborns with BWS do not have macroglossia until closer to their anticipated delivery date.
Given the variation among individuals with BWS and the lack of a simple diagnostic test, identifying BWS can be difficult. In an attempt to standardize the classification of BWS, DeBaun et al. have defined a child as having BWS if the child has been diagnosed by a physician as having BWS and if the child has at least two of the five common features associated with BWS (macroglossia, macrosomia, midline abdominal wall defects, ear creases/ear pits, neonatal hypoglycemia). Another definition presented by Elliot et al. includes the presence of either three major features (anterior abdominal wall defect, macroglossia, or prepostnatal overgrowth) or two major plus three minor findings (ear pits, nevus flammeus, neonatal hypoglycemia, nephromegaly, or hemihyperplasia).
While most children with BWS do not develop cancer, children with BWS do have a significantly increased risk of cancer. Children with BWS are most at risk during early childhood and should receive cancer screening during this time.
In general, children with BWS do very well and grow up to become adults of normal size and intelligence, usually without the syndromic features of their childhood.
Neoplasms
Most children (>80%) with BWS do not develop cancer; however, children with BWS are much more likely (~600 times more) than other children to develop certain childhood cancers, particularly Wilms' tumor (nephroblastoma), pancreatoblastoma and hepatoblastoma. Individuals with BWS appear to only be at increased risk for cancer during childhood (especially before age four) and do not have an increased risk of developing cancer in adulthood. If 100 children with BWS were followed from birth until age ten, about 10 cases of cancer would be expected in the group before age four, and about 1 case of cancer in the group would be expected between age four and ten. In addition to Wilms tumor and hepatoblastoma, children with BWS are also at increased risk of developing adrenal cortical carcinoma, neuroblastoma, and rhabdomyosarcoma.
Both Wilms tumor and hepatoblastoma can usually be cured if diagnosed early. Early diagnosis allows physicians to treat the cancer when it is at an early stage. In addition, there is less toxic treatment. Given the importance of early diagnosis, all children with BWS should receive cancer screening.
An abdominal ultrasound every 3 months until at least eight years of age is recommended and a blood test to measure alpha-fetoprotein (AFP) every 6 weeks until at least four years of age. Families and physicians should determine screening schedules for specific patients, especially the age at which to discontinue screening, based upon their own evaluation of the risk-benefit ratio.
Genetics
Most (>85%) cases of BWS are sporadic, meaning that, typically, no one else in that family has BWS, and parents of an affected child are not at increased risk of having other children with BWS. However, some (<15%) cases of BWS are familial, meaning that a close relative may also have BWS, and parents of an affected child may be at increased risk of having other children with BWS. BWS has been shown to specifically involve mutations in a defined region on the short arm of chromosome 11 referred to as 11p15, that leads to overactivity of the IGF-2 gene (growth factor) and/or no active copy of CDKN1C (inhibitor of cell proliferation gene).
BWS can be caused by a range of different genetic defects. Over five distinct errors involving 11p15 have been identified in different BWS patients. Some patients have maternal chromosomal rearrangements of 11p15. Other patients have paternal uniparental disomy (UPD) of chromosome 11, meaning that the maternal copy of this chromosome is replaced with an extra paternal copy. Many other patients have abnormal DNA methylation in different areas of 11p15, meaning that normal epigenetic marks that regulate imprinted genes in this region are altered. A few other patients have a single gene copy located within 11p15, instead of two copies.
The absence of a mutation in a child with clinical findings suggestive of BWS should not preclude a diagnosis of BWS. Even after extensive molecular testing, the specific defect causing BWS in an affected individual may remain unknown. In 3 out of 10 BWS patients in a small experiment, the genetic or epigenetic mutation could not be found. This fact demonstrates why BWS remains a clinical, rather than genetic, diagnosis, since physicians cannot identify and test for all the genetic causes of BWS. The clinical definition used for BWS is limited, because no standard diagnostic criteria exist that have been independently verified with patients who have either genetic or epigenetic mutations. When molecular analyses were completed in 10 children who met a research criterion for BWS, only 7 of the 10 children had genetic or epigenetic mutations.
Given that the genetics of BWS are complex, a child with BWS should be under the medical care of a geneticist or an expert in the management of BWS.
Genes involved are IGF-2, CDKN1C, H19, and KCNQ1OT1.
Association with CDKN1C
CDKN1C is a protein coding gene that encodes a cyclin-dependent kinase inhibitor that acts as a negative regulator of cell proliferation, effectively making CDKN1C a tumor suppressor gene. CDKN1C also works during fetal development, preventing the fetus from becoming too large. It is located on the short arm of the human chromosome 11 in the ICR2 region, along with many other imprinted genes. Since CDKN1C is preferentially maternally expressed, hypomethylation in the ICR2 region of the maternal allele can result in pathologies such as cancer or a defect known as Beckwith-Weidemann Syndrome. Beckwith-Weidemann Syndrome (BWS) may also be brought about by CDKN1C 11p15 epimutations. It may also be a result of deletions of small amounts of DNA that cause chromosomal abnormalities, rendering the gene inactive. This leaves only the paternally expressed IGF2 to promote cell proliferation. The reduction of growth restriction results in the overgrowth of many tissues, leading to the common symptoms of BWS. These symptoms may include macroglossia, organomegaly, periorbital fullness, and hernias. Knockout models for CDKN1C in mice do exist; in fact, many of the affected offspring exhibit fetal and neonatal lethality and have most of the features related to Beckwith-Weidemann Syndrome.
Management
Abdominal wall defects are common in newborns with BWS and may require surgical treatment. These defects can range in severity from omphalocele (most serious) to umbilical hernia and diastasis recti (least serious). An omphalocele is a congenital malformation in which a newborn's intestines, and sometimes other abdominal organs, protrude out of the abdomen through the umbilicus. Newborns with an omphalocele typically require surgery to place the abdominal contents back into the abdomen in order to prevent serious infection or shock. An umbilical hernia is also a defect in which abdominal contents come through weak abdominal wall muscle at the umbilicus. In general, newborns with umbilical hernias do not require treatment because often these hernias spontaneously close by age four. If, after this time, a hernia is still present, surgery may be recommended. Diastasis recti is a separation of the left and right sides of the rectus abdominis muscle that are normally joined together. Children with diastasis recti usually require no treatment because the condition resolves as the child grows.
Neonatal hypoglycemia, low blood glucose in the first month of life, occurs in about half of children with BWS. Most of these hypoglycemic newborns are asymptomatic and have a normal blood glucose level within days. However, untreated persistent hypoglycemia can lead to permanent brain damage. Hypoglycemia in newborns with BWS should be managed according to standard protocols for treating neonatal hypoglycemia. Usually this hypoglycemia can easily be treated with more frequent feedings or medical doses of glucose. Rarely (<5%) children with BWS will continue to have hypoglycemia after the neonatal period and require more intensive treatment. Such children may require tube feedings, oral hyperglycemic medicines, or a partial pancreatectomy.
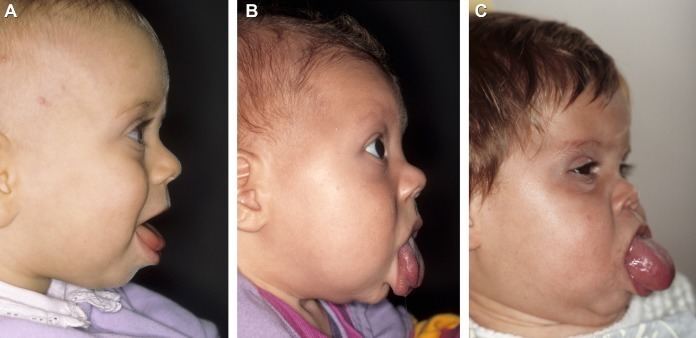
Macroglossia, a large tongue, is a very common (>90%) and prominent feature of BWS. Infants with BWS and macroglossia typically cannot fully close their mouth in front of their large tongue, causing it to protrude out. Macroglossia in BWS becomes less noticeable with age and often requires no treatment; but it does cause problems for some children with BWS. In severe cases, macroglossia can cause respiratory, feeding, and speech difficulties. Children with BWS and significant macroglossia should be evaluated by a craniofacial team.
The best time to perform surgery for a large tongue is not known. Some surgeons recommend performing the surgery between 3 and 6 months of age. Surgery for macroglossia involves removing a small part of the tongue so that it fits within the mouth to allow for proper jaw and tooth development.
Nevus flammeus (port-wine stain) is a flat, red birthmark caused by a capillary (small blood vessel) malformation. Children with BWS often have nevus flammeus on their forehead or the back of their neck. Nevus flammeus is benign and commonly does not require any treatment.
Hemihypertrophy (hemihyperplasia) is an abnormal asymmetry between the left and right sides of the body occurring when one part of the body grows faster than normal. Children with BWS and hemihypertrophy can have an isolated asymmetry of one body part, or they can have a difference affecting the entire one side of the body. Individuals who do not have BWS can also have hemihypertrophy. Isolated hemihypertrophy is associated with a higher risk for cancer. The types of cancer and age of the cancers are similar to children with BWS. As a result, children with hemihypertrophy should follow the general cancer screening protocol for BWS.
Hemihypertrophy can also cause various orthopedic problems, so children with significant limb hemihyperplasia should be evaluated and followed by an orthopedic surgeon.
Hemihyperplasia affecting the face can sometimes cause significant cosmetic concerns that may be addressed by a cranial facial team.
Prognosis
In general, the prognosis is very good. Children with BWS usually do very well and grow up to become the heights expected based on their parents' heights. While children with BWS are at increased risk of childhood cancer, most children with BWS do not develop cancer and the vast majority of children who do develop cancer can be treated successfully.
Children with BWS for the most part had no significant delays when compared to their siblings. However, some children with BWS do have speech problems that could be related to macroglossia or hearing loss.
Advances in treating neonatal complications and premature infants in the last twenty years have significantly improved the true infant mortality rate associated with BWS. In a review of pregnancies that resulted in 304 children with BWS, no neonatal deaths were reported. This is compared to a previously reported mortality rate of 20%. The data from the former study was derived from a BWS registry, a database that may be slightly biased towards involving living children; however, death was not an exclusion criterion to join the registry. This suggests that while infants with BWS are likely to have a higher than normal infant mortality risk, it may not be as high as 20%.
Assisted reproductive technology
Assisted reproductive technology (ART) is a general term referring to methods used to achieve pregnancy by artificial or partially artificial means. According to the CDC, in general, ART procedures involve surgically removing eggs from a woman's ovaries, combining them with sperm in the laboratory, and returning them to the woman's body or donating them to another woman. ART has been associated with epigenetic syndromes, specifically BWS and Angelman syndrome. Three groups have shown an increased rate of ART conception in children with BWS. A retrospective case control study from Australia found a 1 in 4000 risk of BWS in their in-vitro population, several times higher than the general population. Another study found that children conceived by in vitro fertilisation (IVF) are three to four times more likely to develop the condition. No specific type of ART has been more closely associated with BWS. The mechanism by which ART produces this effect is still under investigation.
Epidemiology
Beckwith–Wiedemann syndrome has an estimated incidence of one in 13,700; about 300 children with BWS are born each year in the United States. The exact incidence of BWS is unknown because of the marked variability in the syndrome's presentation and difficulties with diagnosis. The number of reported infants born with BWS is most likely low because many are born with BWS, but have clinical features that are less prominent and therefore missed. BWS has been documented in a variety of ethnic groups and occurs equally in males and females.
Children conceived through In vitro fertilization have a three to fourfold increased chance of developing Beckwith–Wiedemann syndrome. It is thought that this is due to genes being turned on or off by the IVF procedures.
History
In the 1960s, Dr. John Bruce Beckwith, an American pathologist and Dr. Hans-Rudolf Wiedemann, a German pediatrician, independently reported cases of a proposed new syndrome. Originally termed EMG syndrome (for exomphalos, macroglossia, and gigantism), this syndrome over time became known as Beckwith–Wiedemann syndrome or Wiedemann Beckwith syndrome.
Originally, Dr. Hans-Rudolf Wiedemann (born 16 February 1915, Bremen, Germany, died 4 August 2006, Kiel) coined the term exomphalos-macroglossia-gigantism (EMG) syndrome to describe the combination of congenital abdominal wall defects as hernia (exomphalos), large tongues (macroglossia), and large bodies and/or long limbs (gigantism). Over time, this constellation was renamed Beckwith–Wiedemann syndrome following the autoptical observations of Prof. John Bruce Beckwith (born 18 September 1933, Spokane, Washington), who also observed a severe increase in the size of the adrenal glands in some of these patients.